实验和治疗医学
Experimental and Therapeutic Medicine
2013年2月;5 (2):475 - 478。
心脏激素是人类癌细胞分泌卷曲蛋白相关蛋白-3的有效抑制剂
威廉P.斯凯尔顿,四,1,2米歇尔斯凯尔顿,1和大卫L.维斯利1
摘要
分泌卷曲蛋白相关蛋白(sFRPs)是参与肿瘤生长的分泌糖蛋白。心脏合成的四种激素分别是血管扩张器、心钠素肽(ANP)、kaliuretic peptide (KP)和长效利钠肽(LANP),在体内外均具有抗癌作用。我们评估了这些心脏激素在人类胰腺癌、结直肠癌和肾腺癌细胞系中抑制与肿瘤侵袭性相关的sfp -3的能力。血管扩张器、KP、ANP和LANP最大程度地降低了人结直肠腺癌细胞中sFRP-3的浓度,分别为83%、83%、84%和83% (P均<0.0001)。在人胰腺癌细胞中,血管扩张器、KP、ANP和LANP治疗后,sfp -3的浓度最大降低77%、77%、77%和78% (P均<0.0001)。在人肾腺癌细胞中,血管扩张器、KP、ANP和LANP对sFRP-3的降低最大分别为68%、66%、68%和66% (P<0.0001)。结果表明,这四种心脏激素是重要的抑制剂(高达84%)的sFRP-3在各种人类癌细胞。此外,这些数据表明,心脏激素对sfp -3的代谢靶向作用有助于其抗癌作用机制。
关键词:分泌卷曲蛋白相关蛋白3、心脏激素、结直肠腺癌、胰腺癌、肾腺癌
介绍
分泌卷曲蛋白相关蛋白(sFRPs)是棕榈酰化的分泌糖蛋白,参与细胞增殖和肿瘤生长(1,2)。sFRPs由约300个氨基酸组成,其氨基末端由富含半胱氨酸结构域(CRD)组成,与卷曲受体的活性位点具有30-50%的同源性(3,4)。frizzle的CRD是Wnt结合和随后信号转导的活性位点。这类CRD守恒在不同的蛋白质,包括seven-transmembrane类的受体酪氨酸激酶受体酪氨酸kinase-like孤儿受体(ROR)家庭(5)。sFRPs作为细胞外信号配体,形成一个能够表达下调Wnt信号抑制与卷曲的受体(6)复杂。因为Wnt导致癌细胞生长,最初假设sFRPs是癌症细胞生长抑制剂(7),但随后的研究表明sFRP-3(也称为FrzB)存在高水平的转移性肾癌组织(8)。研究还表明,sFRP-3促进入侵肾癌细胞(8)。sFRPs与其他类型的癌症肿瘤促进活动(9)。sFRP-3水平升高的各种类型的癌症表明,它可能是一个有价值的目标(7)治疗。
四个内生心脏激素(血管扩张器,kaliuretic肽(KP)、心房利钠肽(ANP)和长效利钠肽(LANP)]有抗癌作用体内(10 - 12)和体外已经报告给人类肾癌细胞的数量减少了81%(13),人类大肠癌细胞的89 - 97%(14)和胰腺癌细胞65% (15)。本研究旨在探讨四种心脏激素在人肾癌、人胰腺癌和人结直肠癌细胞中是否抑制sfp -3的作用机制。结果表明,这四种心脏激素中的每一种都能有效地抑制三种不同类型癌症中的sFRP-3Cardiac hormones are potent inhibitors of secreted frizzled-related protein-3 in human cancer cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3570200/
胰岛素抵抗与心力衰竭:分子机制Insulin Resistance and Heart Failure: Molecular Mechanisms
胰岛素抵抗和与之相关的心脏胰岛素代谢信号的减少正在成为心力衰竭发展的一个主要因素,并且由于肥胖症、心肾代谢综合征和我们的老龄化人口的流行增加而变得更为重要。导致心脏胰岛素抵抗的主要因素是氧化应激、高血糖、高脂血症、脂肪因子/细胞因子分泌失调、肾素-血管紧张素-醛固酮系统(RAAS)和交感神经系统的不适当激活。与全身胰岛素抵抗相关的代谢、内分泌和细胞因子改变会加剧心脏胰岛素抵抗的影响。这些不同变化的集合导致胰岛素抵抗表型,代谢不稳定、钙处理受损、线粒体功能障碍和氧化应激、心肌内皮相互作用失调导致能量不足、舒张功能受损、心肌细胞死亡和心肌纤维化。因此,了解胰岛素抵抗与心力衰竭之间的分子机制有助于设计新的、更有效的机制药物来改善心肌和全身胰岛素抵抗。
关键词:心脏胰岛素抵抗,心肾代谢综合征心力衰竭(HF)是西方世界发病率和死亡率的主要原因。高频出现在超过500万人在美国,占每年近400亿美元的医疗保健费用(1)冠心病(CHD),糖尿病,高血压,和心肺肾综合症(CRS)是导致心力衰竭的主要因素(1、2)。CRS是一个星座的代谢紊乱,包括肥胖、高血压、胰岛素抵抗、心肌和肾功能异常。CRS会增加患2型糖尿病(T2DM)、肾脏和心血管疾病(CVD)、冠心病和HF的风险(3)。由于人口老龄化、营养过剩和久坐不动的生活方式的流行,CRS的发病率正在以惊人的速度增长。流行病学研究表明,超过60%的美国成年人超重,超过40%的60岁以上的美国人患有CRS。此外,儿童-成人肥胖也正在成为一个全球性的健康问题,容易导致CRS和T2DM(1-4)。
1. 鸡和蛋:胰岛素抵抗和心力衰竭
胰岛素抵抗导致心肌病
胰岛素抵抗与心力衰竭之间存在着很强的相关性。胰岛素抵抗预测心力衰竭的发展几个临床研究(5)。尽管胰岛素抵抗协会心力衰竭可能归因于CRS等条件有关,高血压、冠心病、心力衰竭的识别与糖尿病没有冠心病和高血压(糖尿病心肌病),和肥胖没有糖尿病,高血压和冠心病(肥胖性心肌病)提出了一个有趣的假设,即胰岛素抵抗本身对心功能有深远的负面影响(6,7)。重型饮酒与胰岛素抵抗的形成和心脏功能障碍(酒精性心肌病)(8)。胰岛素抵抗之间的因果联系和心脏功能障碍在胰岛素抵抗的形成由遗传和环境因素(9)转基因小鼠cardiomyocyte-specific删除IR (CIRKO)或胰岛素受体底物(CIRSKO)刺激葡萄糖摄取减少,心脏也有障碍函数。此外,葡萄糖转运体4 (GLUT4) KO小鼠也会发生心功能障碍,从而提示胰岛素抵抗是CRS收缩功能障碍发生的一个促进因素(10-12)。
心力衰竭导致胰岛素抵抗
HF的存在可以预测胰岛素抵抗的发展,HF患者每10年患T2DM的风险比高血压患者高18%到22%。28%的老年心力衰竭患者在3年以上发展为糖尿病,在多因素分析中,充血性心力衰竭预测发展为2型糖尿病。心力衰竭患者可能同时具有全身性和心脏性胰岛素抵抗。心肌梗死后也可见胰岛素抵抗(13-15)。
2. 胰岛素信号及其在心脏和血管系统中的调节
胰岛素信号在心脏和血管系统
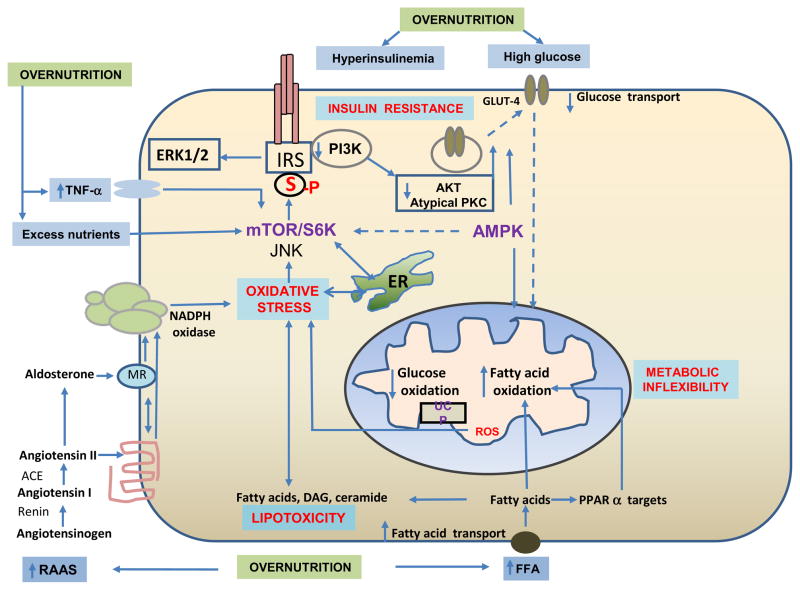
胰岛素信号通路有两种:磷脂酰肌醇3激酶(PI3- kinase)/蛋白激酶B (Akt)信号通路主要诱导代谢反应,而丝裂原活化蛋白激酶(MAPK)信号通路主要诱导生长因子样反应(16,17)。在这一路径选择信号(图1)中,配体激活的胰岛素受体的激活磷酸化了胰岛素受体底物1 (IRS-1)。IRS-1上酪氨酸残基的磷酸化导致Src同源性2 (SH2)与SH2域信号分子(包括pi3 -激酶和生长因子受体结合蛋白2 (GRB2)的域结合基序的参与。当PI3- kinase的p85调控亚基SH2结构域与IRS-1上的酪氨酸磷酸化基序结合时,激活预先相关的p110催化亚基,生成磷脂酰肌醇(3,4,5)三磷酸(P3)。这个分子然后结合pleckstrin同源域3-phosphoinositide端依赖蛋白激酶1 (PDK-1),导致其磷酸化和其他下游激酶包括一种蛋白激酶的激活和非典型的蛋白激酶C (PKC)亚型,调解的行为包括GLUT-4膜易位,导致心肌组织葡萄糖摄取和骨骼肌,一氧化氮(NO)介导的冠状血管舒张,代谢的灵活性,以及能量稳态(16,17)。MAPK信号通路包括酪氨酸磷酸化IRS-1或Src同源性2 -含有蛋白质和胶原蛋白同源区域(自燃)绑定的SH2域GRB2导致激活pre-associated三磷酸鸟苷交换因素son-of-sevenless (SOS)和GTP-binding蛋白质rat-sarcoma (RAS)磷酸化/激活细胞外signal-regulated激酶(ERK) 1/2。这些信号通路促进生长和重构反应,并由此导致心肌肥厚、心肌纤维化、心肌内皮信号受损以及心肌和内皮细胞死亡。蛋白质酪氨酸磷酸酶或脂质磷酸酶对磷酸化的调控提供了一个负调控环(16-18)。
图1
营养过剩和胰岛素代谢信号受损导致心肌病
引起胰岛素抵抗的主要信号通路是多种刺激的聚合导致S6激酶的激活。营养过剩导致循环脂肪酸、氨基酸和葡萄糖水平升高,从而激活mTOR/S6激酶。RAAS的激活导致氧化应激,这是由于Ang II和醛固酮激活NADPH氧化酶所致。氧化应激可激活JNK、S6激酶等激酶,导致IRS-1丝氨酸磷酸化,抑制胰岛素代谢信号。细胞因子诱导的胰岛素抵抗也包括激活S6激酶。氧化应激和胰岛素抵抗还伴有AMPK信号通路受损,AMPK信号通路是mTOR/S6激酶的负调控因子。mTOR/S6激酶的持续高活化也会导致内质网应激、氧化应激和JNK的活化。脂肪酸的过量积累和氧化作用的增强伴随着脂质中间体积累的增加,导致脂肪毒性和代谢不灵活性。这四种营养过剩引起的细胞事件通过它们的前馈相互作用被进一步放大。
胰岛素信号调节
胰岛素信号通路中促进胰岛素抵抗的主要汇聚点是对接蛋白-1。IRS-1的丝氨酸残基被多种激酶磷酸化,包括蛋白激酶C (PKC)、C- jun n -末端激酶(JNK)、哺乳动物雷帕霉素靶蛋白(mTOR)和核糖体p70 S6激酶1 (S6K1),是调控IRS-1功能的主要机制(16-18)。IRS-1丝氨酸残基磷酸化减弱IRS-1酪氨酸磷酸化及其与PI-3激酶p85亚基的关联,并触发其蛋白酶体依赖降解。蛋白酶体的降解可以通过抑制细胞因子信号转导(SOCS)-3介导但与磷酸化无关的机制发生。不适当的激活酪氨酸或脂质磷酸酶,Akt FOXO转录因子,AMP -激活的蛋白激酶(AMPK)信号抑制mTOR / S6激酶,监管IRS-1表达式和IRS-1信号组件的微rna,并增加表达毛球族同族体蛋白3 (TRIB3)调制IRS-1酪氨酸磷酸化和Akt激活也导致受损的胰岛素信号和胰岛素抵抗(16号)
3.心脏胰岛素抵抗的分子机制
心脏胰岛素抵抗的发生可能与全身胰岛素抵抗无关,但全身胰岛素抵抗对心脏胰岛素抵抗有重要作用,其次是营养循环水平的增加、氧化应激、神经体液和细胞因子平衡的改变(图1)。
营养过剩
外周胰岛素抵抗或营养过剩引起的循环游离脂肪酸(FFA)和甘油三酯水平增加,并通过增加摄取在心肌细胞中积累。其结果是脂质分子如二酰基甘油(DAG)、脂肪酸和神经酰胺的积累增加,这些分子通过激活导致IRS-1丝氨酸磷酸化增加的激酶而导致胰岛素抵抗。此外,高血糖诱导氧化应激,进而激活氧化还原敏感激酶和增加IRS-1磷酸化。高血糖引起的氧化也会引起蛋白激酶C的激活和心脏内血管紧张素II (Ang II)信号的上调。过量的葡萄糖和氨基酸也会激活mTOR/S6K1。高胰岛素血症单独引起胰岛素抵抗,并进一步增强高糖和棕榈酸诱导的胰岛素抵抗(16-22)。
发病
腹部肥胖的中心特征可能会导致CRS的其他特征及其并发症的发展,这些特征可能会在以后发展(16-18)。除了增加远期运费协议的版本,脂肪细胞特异表达功能和巨噬细胞激活导致增加分泌细胞因子,如肿瘤坏死因子(TNF) -α和白介素6 (il - 6)和抵抗素等发病而降低脂联素的分泌。TNF-α通过激活MAPK和il - 6引起胰岛素抵抗,PKC mTOR / S6K1和IRS-1 SOCS-3介导蛋白酶体降解。抵抗素诱导胰岛素抵抗和炎症,而脂联素改善胰岛素抵抗(3,12,16,17,23 - 25)。
肾素-血管紧张素-醛固酮(RAAS)和交感神经系统(SNS)的激活
RAAS和SNS的激活不仅见于与胰岛素抵抗相关的心力衰竭早期,也见于慢性心力衰竭或心肌梗死(24- 27)。除醛固酮的合成和二世的机制导致了醛固酮与胰岛素抵抗之间的联系包括触发醛固酮释放肾上腺肿瘤坏死因子α等细胞因子,il - 6和释放脂肪组织的脂质因素。醛固酮和Ang II均可激活血管平滑肌细胞、心脏和骨骼肌组织中膜结合NADPH氧化酶复合物,产生活性氧(ROS)。Ang - II与醛固酮之间的非基因组效应的正交叉作用导致活性氧产生的增强效应。ROS的产生进而导致氧化还原敏感激酶的激活,如蛋白S6激酶、蛋白激酶C同工酶和丝裂原活化蛋白激酶;从而导致IRS-1丝氨酸磷酸化导致胰岛素抵抗。最近的研究提示RAAS与哺乳动物雷帕霉素复合物I (mTORC1)/S6K1靶蛋白在血管紧张素2型受体(AT2R)介导下的活化相互作用,可能存在mTOR-AT2R信号通路的适应性作用(28)。虽然交感神经系统的异常激活是胰岛素抵抗和心力衰竭的另一个组成部分,但它通常与RAAS的激活有关(26,27)。
线粒体氧化应激和内质网应激
线粒体是细胞ROS的主要来源,发生这种情况(1)继发于细胞氧化应激,涉及NADPH氧化酶或黄嘌呤氧化酶,(2)线粒体电子传递失调,(3)线粒体NADPH氧化酶4 (NOX4)表达增加,(4)蛋白赖氨酸乙酰化改变(27,28)。线粒体氧化应激已被证明通过IRS-1丝氨酸磷酸化破坏胰岛素信号通路(27,28)。内质网应激可导致线粒体氧化应激和胰岛素抵抗。虽然内质网应激诱导胰岛素抵抗的机制尚不清楚,但应激激活的MAP激酶JNK的激活被认为是内质网应激与胰岛素抵抗之间的信号通路之一(28,29)。
4. 胰岛素抵抗背景下心力衰竭的分子机制
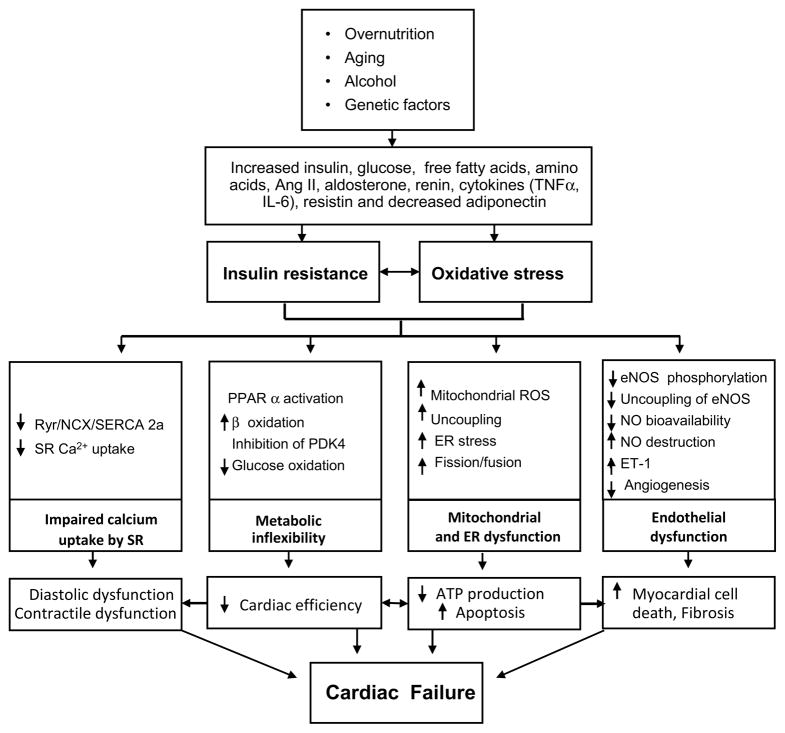
心力衰竭与左室肥厚、左室质量指数增加、心肌细胞死亡、扩张型心肌病、细胞外纤维化和影响舒张和收缩功能的功能异常有关。心功能障碍的发生既有心血管胰岛素抵抗的结果,也有周围和肝脏胰岛素抵抗的结果(5,9,14,18,22)。导致心脏损伤的因素包括钙信号受损、底物代谢改变、线粒体功能障碍和氧化应激、内质网应激以及心肌内皮细胞相互作用失调。其结果是钙处理和收缩能力受损,心脏能量效率降低,心肌细胞死亡和心肌纤维化(图2)。
图2
心脏胰岛素抵抗的分子机制以及胰岛素信号通路受损和营养过剩导致的心功能障碍
胰岛素抵抗和氧化应激导致心脏功能障碍的主要后果是钙处理功能受损、代谢不灵活、线粒体和ER功能障碍以及内皮细胞功能障碍。这些事件导致心脏功能低下、心肌细胞死亡、心肌纤维化和舒张功能障碍。心脏损伤的进展导致收缩功能障碍和心力衰竭。
钙处理受损
细胞内钙在调节心功能中起着至关重要的作用。钙诱导的钙释放通过激活离子通道、激活ryanodine受体(Ryr)和钠钙交换器(NCX)活性来调节心肌收缩力。心肌舒张主要通过肌浆内质网Ca2+ - atp酶2a (SERCA 2a)的活性在肌浆网Ca2+ -ATPase 2a的重新螯合而发生。舒张功能受损是CRS和T2DM中发现的最早的心功能障碍。在CRS和糖尿病心肌病中,Ryr受体、SERCA2a和NCX的表达或活性异常,肌浆网钙摄取受损。pi3 -激酶/Akt信号通路已被证明可调节细胞内钙离子,彻底增强l型钙通道功能,上调SERCA2a的表达和活性,而心脏胰岛素抵抗减弱pi3 -激酶/Akt信号通路,从而导致钙处理受损(3,6,30 - 35)。
底物代谢改变和心脏效率受损
底物代谢的早期异常
减少葡萄糖氧化主要发生由于葡萄糖减少条目虽然GLUT-4和增加脂肪酸积累通过抑制葡糖激酶和丙酮酸脱氢酶(3)。增加脂肪酸的通量成心肌细胞引起的系统性/脂肪组织胰岛素抵抗(IR)和IR-associated再分配集群分化蛋白36 (CD36)质膜导致增加脂肪酸氧化。在这种情况下,心脏的表达过氧物酶体扩散者激活受体α(PPAR-α)增加。此外,ppar被包括脂肪酸在内的配体激活。一旦激活,PPAR-α提高蛋白质的转录控制脂肪酸吸收(脂蛋白脂肪酶,CD36和脂肪酸结合蛋白),和氧化(中、长链酰基辅酶a脱氢酶和羟基酰基辅酶a脱氢酶)。由于葡萄糖是一种更有效的底物,心脏代谢从葡萄糖代谢转换到脂肪酸氧化降低了心脏效率。这将导致心脏衰竭的代谢压力进一步增加。通过围产期谷氨酰胺-4葡萄糖转运体的表达来预防db/db小鼠底物代谢的改变,可以预防db/db小鼠的心功能障碍,这进一步有利于与T2DM相关的胰岛素抵抗中代谢开关的存在(34-35)。
底物代谢的晚期改变和失代偿性心力衰竭
随着病情的进展,心肌呈胎儿基因计划表型,调节脂肪酸-氧化的基因表达下调,从而进一步降低心肌的代谢效率。在这种情况下,AMPK信号受损,过氧物酶体扩散者激活受体γco-activator(包括)1α是衰减和PPARα表达式(到三十五)有所下降。
心肌损伤和胰岛素抵抗叠加
另一个日益被认识到的使临床管理具有挑战性的情况是,在心肌梗死或长期心力衰竭和缺血性心脏病后,胰岛素抵抗的发展伴随着RAAS、SNS和氧化应激的激活。在这些条件下,胰岛素抵抗的存在破坏了心肌对葡萄糖氧化的适应,而葡萄糖氧化是心肌存活所必需的;导致心肌细胞死亡增加,预后不良(9,14,15,33 - 35)。
线粒体功能障碍、氧化应激和内质网应激
与线粒体动力学改变(分裂和融合)相关的心肌线粒体生物发生的减少和增加,已在人和动物中观察到,CRS与ATP水平降低和线粒体电子传输功能障碍同时发生。这些不同的发现要么与CRS的持续时间有关,要么与相关的潜在疾病有关。线粒体氧化应激和内质网应激也促进心肌细胞凋亡(27,28,36,37)。心肌自噬在心功能障碍中的作用目前尚不清楚(38,39)。
心肌细胞内皮一氧化氮(NO)信号受损
内皮功能障碍是胰岛素抵抗与心力衰竭的重要联系。内皮细胞的胰岛素抵抗导致一氧化氮或一氧化氮合酶解耦联受损。这导致缺氧和抑制血管生成导致心肌细胞死亡。内皮细胞胰岛素抵抗还导致内皮素-1(ET-1)释放增加,从而导致心肌肥厚和纤维化(3,6,7,40)。
至:
5. 针对心力衰竭患者的胰岛素抵抗
生活方式干预
慢性心力衰竭患者的胰岛素抵抗的临床意义往往代表了这些个体中潜在的可逆代谢紊乱,尤其是在心功能障碍的早期阶段。生活方式干预,如减少热量摄入和酒精摄入,增加锻炼,似乎可以改善全身和组织的胰岛素抵抗。这些措施针对几个与胰岛素抵抗有关的胰岛素信号级联,包括抑制TRIB3表达,从而克服TRIB3介导的胰岛素抵抗(3,5,21,41)。
胰岛素抵抗
除了改善肝脏和外周胰岛素抵抗,二甲双胍治疗糖耐量受损在动物模型中已被证明可以逆转心室功能障碍。在胰岛素抵抗心肌细胞中,二甲双胍促进葡萄糖转运体1 (GLUT-1)和GLUT-4向肌膜转移。它以ampk依赖的方式促进葡萄糖摄取,并通过减少细胞内Ca瞬变,防止高糖诱导的弛豫异常。二甲双胍增加脂肪酸氧化(FAO),但对葡萄糖代谢和AMPK活化的影响似乎维持代谢的灵活性(9,13,42 - 43)。
激活RAAS和SNS
鉴于胰岛素抵抗作为HF患者死亡的独立危险因素的重要性,血管紧张素转换酶抑制剂(ACEIs)和血管紧张素受体阻滞剂(ARBs)的使用已被报道可在HF患者的大型试验中显著降低新发糖尿病的发生率。ACEIs和ARBs在改善胰岛素抵抗或高血压方面并不完全有效,醛固酮逸出常常阻碍它们的疗效(26,44,45)。直接肾素抑制剂aliskiren是一种有效的选择性肾素抑制剂(26),该酶被认为是RAAS的限速步骤(26,44)。肾素抑制与Ang II水平的降低和血压的降低有关(46,47)。当MR拮抗剂(MRAs)与ACEIs、ARBs和β-受体阻滞剂联合使用时,还可改善左心室功能和临床结果。然而,一些临床研究将MR拮抗剂与异常的葡萄糖稳态联系起来,从而提示了MR受体信号的复杂性(26,44,45)。非选择性受体阻滞剂不会恶化胰岛素敏感性,如奈比沃洛尔和卡维地洛尔,似乎在HF中发挥有益的作用(48)。Nebivolol还能抑制Ang ii诱导的NADPH氧化酶和ROS的激活,从而为对胰岛素敏感性产生有益影响提供了理论依据(49)。
底物代谢
调节胰高血糖素样肽1 (GLP-1)信号通路的药物包括GLP-1 mimetics或二肽二肽酶(DPP)-4抑制剂,据报道,这些药物通过刺激胰腺分泌胰岛素来控制高血糖,并通过谷氨酰胺-1和谷氨酰胺-4转运到肌膜来提高心肌葡萄糖摄取(9,50)。慢性输注GLP-1除了改善心脏胰岛素敏感性外,还可改善左心室功能障碍、运动耐受性和生活质量。DPP-4抑制剂西格列汀改善了冠心病患者的左心室功能,减轻了缺血后昏迷,保留了左心室射血分数(LVEF)。利纳格列汀也被证明对2型糖尿病患者有心血管(CV)益处(9,50,51)。曲美他嗪通过抑制心肌底物的氧化磷酸化,通过抑制长链3-酮酰基辅酶a(β-氧化中的最后一种酶),将代谢从游离脂肪酸转移到葡萄糖氧化,从而影响心肌底物的利用。在小型临床研究中,该药在降低脂肪酸氧化、改善心室功能和降低心力衰竭患者的胰岛素抵抗方面有微小但静态的显著改善(13,33,52,53)。
线粒体氧化应激和内质网应激
针对线粒体氧化应激和内质网应激的新化合物正在开发中。在这方面,合成的Szezo-Schiller肽S-31靶向线粒体的产生,在Ang II模型和高血压性心肌病模型中预防心肌肥厚和纤维化,改善左室舒张功能(54)。fda批准的药物已经被证明可以减少ER压力,在动物实验中也可以改善心功能,这表明它们可能用于抑制人类胰岛素抵抗(55)。
至:
总结
胰岛素抵抗与HF密切相关。它与HF的主要因素有关。在临床情况下,这两种情况通常共存,但胰岛素抵抗的存在对心力衰竭的进展有不利影响。心血管胰岛素抵抗也受全身胰岛素抵抗的调节。导致胰岛素抵抗的信号通路主要集中在IRS-1。除了糖毒性和脂肪毒性外,神经体液和细胞因子失衡的失调和氧化应激是导致心脏胰岛素抵抗和心功能受损的主要原因。虽然靶向RAAS-SNS系统显著改善了临床结果,但靶向代谢途径、GLP-1信号转导、线粒体功能障碍和ER应激的药物可能在潜在代谢异常的基础上显示出额外的好处。因此,确定基于机制的新的治疗靶点来改善胰岛素抵抗,有望更好地管理胰岛素抵抗和心力衰竭。
要点
在西方工业化国家,心力衰竭与老龄化和糖尿病同时增加。
胰岛素抵抗与心肾代谢综合征和2型糖尿病的舒张功能障碍有关。
胰岛素代谢信号转导是重要的心脏代谢灵活性和缺血后恢复,而信号转导障碍损害这两个过程。Insulin Resistance and Heart Failure: Molecular Mechanisms
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3457065/
2012年8月14日(4):360-5。doi: 10.1007 / s11906 - 012 - 0279 - 2。
心肾代谢综合征中的氧化应激
摘要
过多的内脏脂肪导致肾素-血管紧张素-醛固酮系统的不适当激活,尽管体积膨胀和盐潴留的状态有助于亚临床前氧化剂机制的升高。这些副作用是由活性氧(ROS)的过量产生和抗氧化防御机制的减弱所介导的。多余的组织(即。自由基导致胰岛素依赖的代谢信号通路受损,而胰岛素依赖的代谢信号通路调节葡萄糖的利用/处理和全身胰岛素敏感性。ROS的产生是正常细胞信号和生理反应所必需的。氧化还原稳态的丧失导致促炎/促纤维化的环境,促进胰岛素代谢信号的损伤,内皮介导的血管舒张功能降低,以及相关的心血管和肾脏结构和功能异常。这些不适应过程在高血压的心肾代谢表型进展中越来越被认为是重要的。越来越多的证据支持Ang II信号通过AT(1)R和醛固酮通过MR的作用,与氧化还原介导的内皮、心脏和肾脏功能受损的改变一起,在这一代谢表型中发挥重要作用。有新的临床数据表明,以肾素血管紧张素-醛固酮系统(RAAS)为靶点的治疗也能减轻氧化应激,改善内皮、心脏和肾脏功能,共同有助于降低高血压。Oxidative stress in the cardiorenal metabolic syndrome. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/22581415