��
ȱ����Ӧ�Բ��ԣ�HIF)-����-NDRG3�ź�ͨ· Hypoxia Adaptive Strategy-Lactate-NDRG3 Pathway
��
Hydroxylation of HIF-1: Oxygen Sensing at the Molecular Level
Gregg L. Semenza 2004
Abstract
The ability to sense and respond to changes in oxygenation represents a fundamental property of all metazoan cells. The discovery of the transcription factor HIF-1 has led to the identification of protein hydroxylation as a mechanism by which changes in Po2 are transduced to effect changes in gene expression. Multicellular life on Earth is based on the use of O2 for the efficient generation of high-energy compounds, and O2 consumption increases with the mass and metabolic activity of the organism. However, exposure to O2 must be limited due to the damaging effects of reactive oxygen species (ROS) on cellular macromolecules. Thus all of the major physiological systems of mammals participate in complex homeostatic mechanisms that are designed to maintain the O2 concentration to which each cell is exposed within a narrow range (FIGURE 1). The study of these systems has occupied physiologists for centuries. During the course of the past century, these studies have been extended to the cellular level. Finally, research over the past decade has produced dramatic insights into the molecular mechanisms underlying oxygen homeostasis during both prenatal and postnatal life.
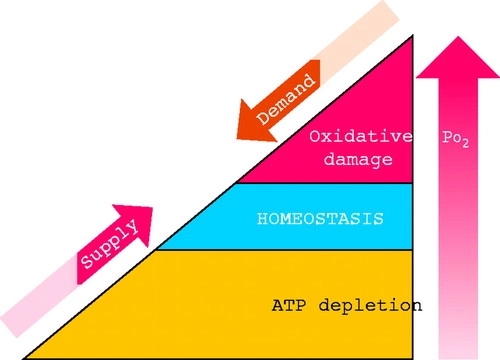
FIGURE 1.Oxygen homeostasis All of the major physiological systems participate in complex homeostatic mechanisms that regulate O2 supply and demand to maintain cellular oxygenation within a narrow range that balances the risks associated with O2 deficiency and excess.
��
Control of Oxygen-Regulated Gene Expression by HIF-1 Physiological responses involve changes in gene expression. The blood O2-carrying capacity is maintained by the O2-regulated production of erythropoietin (EPO), which stimulates the proliferation and survival of red blood cell progenitors. Analysis of cis-acting sequences required for increased transcription of the EPO gene in response to hypoxia led to the identification (70), biochemical purification (81), and molecular cloning (79) of hypoxia-inducible factor-1 (HIF-1). EPO is produced primarily within a rare cell type in the kidney. However, HIF-1 is expressed in all cell types and functions as a master regulator of oxygen homeostasis by playing critical roles in both embryonic development and postnatal physiology. HIF-1 has been identified in all metazoan species that have been analyzed from Caenorhabditis elegans to Homo sapiens (organ-isms whose cell numbers differ by more than 10 orders of magnitude), suggesting that the appearance of HIF-1 represented an adaptation that was essential to metazoan evolution. The expression of over 70 genes is known to be activated at the transcriptional level by HIF-1, and specific HIF-1 binding sites have been identified for many of these genes. Although the list of HIF-1 target genes is extensive (FIGURE 2), it probably underestimates the total number of genes regulated by HIF-1 by at least an order of magnitude. The battery of genes regulated by HIF-1 is different in each cell type, and, for some genes, expression can be induced or repressed by HIF-1 depending on the cell type (34).Among the critical physiological processes regulated by HIF-1 target genes are erythropoiesis, angiogenesis, and glycolysis, which are examples of systemic, local tissue, and intracellular adaptive responses to hypoxia, respectively (30).
��
FIGURE 2.Representative HIF-1 target genes Hypoxia inducible factor-1 (HIF-1) activates the transcription of genes encoding secreted signaling proteins, including angiogenic growth factors and survival factors, cell surface receptors, extracellular matrix proteins and modifying enzymes, transcription factors, cytoskeletal proteins, proapoptotic proteins, and glucose transporters and glycolytic enzymes. ADM, adrenomedullin; ADRA1B, ��1B-adrenergic receptor; ALD, aldolase; ANGPT, angiopoietin; CITED, CREB binding protein (CBP)/p300-interacting transactivator; COL5A1, collagen V ��1-subunit; CTSD, cathepsin D; CXCR, chemokine receptor; DEC, differentiated embryo chondrocyte expressed; EDN, endothelin; ENO, enolase; EPO, erythropoietin; ETS, erythroblastosis virus transforming sequence; FN, fibronectin; GLUT, glucose transporter; GPI, glucose phosphate isomerase; HK, hexokinase; KRT, keratin; LDHA, lactate dehydrogenase A; LEP, leptin; MMP, matrix metalloproteinase; NIP, BCL2/adenovirus E1B 19-kDa-interacting protein; NIX, NIP3-like; P4HA1, prolyl-4-hydroxylase ��1-subunit; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3; PFKL, phosphofructokinase L; PGF, placental growth factor; PGK, phosphoglycerate kinase; PLAUR, urokinase-type plasminogen activator receptor; PROK, prokineticin (endocrine gland-derived VEGF); STC, stanniocalcin; TF, transferrin; TFRC, transferrin receptor; TGFA and TGFB, transforming growth factor-�� and -��; TPI, triose phosphate isomerase; VEGFR, VEGF receptor; VIM, vimentin.
HIF-1 is a heterodimeric protein that is composed of HIF-1�� and HIF-1�� subunits. The amino-terminal half of each subunit consists of basic helix-loop-helix and Per-ARNT-Sim (PAS) domains that mediate heterodimerization and DNA binding. The carboxy-terminal half of HIF-1�� contains two transactivation domains that mediate interactions with coactivators such as CREB binding protein (CBP) and p300 (30, 31, 59). Coactivators interact with both sequence-specific DNA binding proteins such as HIF-1 and with the general transcription factors associated with RNA Polymerase II (reviewed in Ref. 69). Coactivators also have histone acetyltransferase activity that is required to make the DNA embedded in chromatin accessible to the polymerase complex for transcription into RNA. The HIF-1�� subunit is constitutively expressed, whereas the expression and activity of the HIF-1�� subunit are precisely regulated by the cellular O2 concentration. HIF-1�� accumulates instantaneously under hypoxic conditions and on reoxygenation is rapidly degraded, with a half-life of <5 min in posthypoxia tissue culture cells (28, 79). This represents an overestimate of the half-life, because it includes the time required for O2 to diffuse out of the culture medium. In an isolated, perfused, and ventilated lung preparation subjected to hypoxia and reoxygenation, the half-life of HIF-1�� is <1 min (85). No protein has been shown to have a shorter half-life.
In addition to HIF-1��, a structurally and functionally related protein designated HIF-2��, which is the product of the EPAS1 gene, can also heterodimerize with HIF-1�� (77). HIF-1��:HIF-1�� and HIF-2��:HIF-1�� heterodimers appear to have overlapping but distinct target gene specificities (22, 73). Unlike HIF-1��, HIF-2�� is not expressed in all cell types, and when expressed it can be inactive as a result of cytoplasmic sequestration (56). A third protein, designated HIF-3��, has also been identified (18). Its role has not been well defined, although a splice variant, designated IPAS, has been shown to bind to HIF-1�� and inhibit its activity (45, 46). Molecular Mechanisms of Oxygen Sensing The mechanism underlying the dramatic regulation of HIF-1�� protein expression was a source of great debate, with several models proposed that invoked, for example, the functioning of an O2-binding hemoprotein or an ROS-generating NADPH oxidase as central to the oxygen sensing that determined HIF-1�� levels (reviewed in Ref. 66). Among the observations used to support these models was the finding that HIF-1 DNA binding activity and target gene expression were induced in cells exposed to the iron chelator desferrioxamine or to cobalt chloride (80). Remarkably, HIF-1�� transactivation domain function is also induced in cells exposed to hypoxia, iron chelation, or cobalt chloride (30, 31, 59), suggesting a common mechanism for regulating both HIF-1�� expression and activity. The O2-dependent degradation of HIF-1�� involves ubiquitination and degradation by the 26S proteasome (23, 32, 61). The von Hippel-Lindau tumor suppressor protein (VHL) is required for this process (FIGURE 3), because renal carcinoma cells lacking functional VHL constitutively express HIF-1�� and HIF-1 target genes under nonhypoxic conditions (6, 49).VHL forms a complex with elongin B, elongin C, cullin 2, and RBX1 to form an E3 ubiquitin-protein ligase capable of functioning with E1 ubiquitin-activating and E2 ubiquitin-conjugating enzymes to mediate the ubiquitination of HIF-1�� (33).
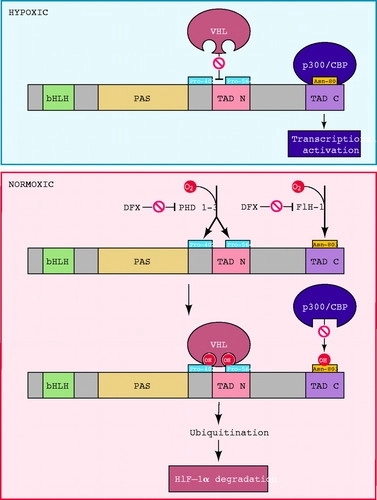
FIGURE 3.Oxygen sensing by hydroxylation of HIF-1�� The amino-terminal half of HIF-1�� consists of basic helix-loop-helix (bHLH) and Per-ARNT-Sim homology (PAS) domains. The carboxy-terminal half contains the transactivation domains (TAD-N and TAD-C). The HIF-1�� prolyl hydroxylases (HPH)/prolyl hydroxylase domain proteins (PHD) 1�C3 hydroxylate Pro-402 and Pro-564. Factor inhibiting HIF-1 (FIH-1) hydroxylates Asn-803. Proline hydroxylation is required for the interaction of HIF-1�� with the von Hippel-Lindau tumor-suppressor protein (VHL), which is the recognition component of an E3 ubiquitin-protein ligase that targets HIF-1�� for proteasomal degradation. Asparagine hydroxylation prevents the interaction of HIF-1�� with the coactivators CBP and p300. The enzymes, which contain Fe(II) at the active site, can be inactivated by desferrioxamine (DFX) and other iron chelators. O2 appears to be a rate-limiting substrate for the hydroxylases under physiological conditions, thus providing a mechanism for the direct regulation of the stability and activity of HIF-1�� as a function of the cellular O2 concentration.
��
A region of HIF-1�� encompassing amino acid residues 400�C600 is necessary and sufficient for O2-regulated ubiquitination and degradation (23, 32, 74). VHL interacts, via its ��-domain, with amino acid residues 532�C585 of HIF-1�� (55, 75). Because the ubiquitination and degradation of other key regulatory proteins such as I��B are regulated by phosphorylation, great effort was made to identify phosphorylatable (serine, threonine, tyrosine) residues of HIF-1�� that were important for regulation of protein half-life, but to no avail. Instead, Pro-564 is hydroxylated in an O2-dependent manner, and this modification is required for VHL binding (25, 27, 87). Pro-402 represents a second site of hydroxylation and VHL binding (48). Pro-402 and Pro-564 are each contained within a similar amino acid sequence (LXXLAP, where A is alanine, L is leucine, P is proline, and X is any amino acid). HIF-2�� and HIF-3�� expression are also regulated by prolyl hydroxylation and VHL binding (20, 49, 50).
��
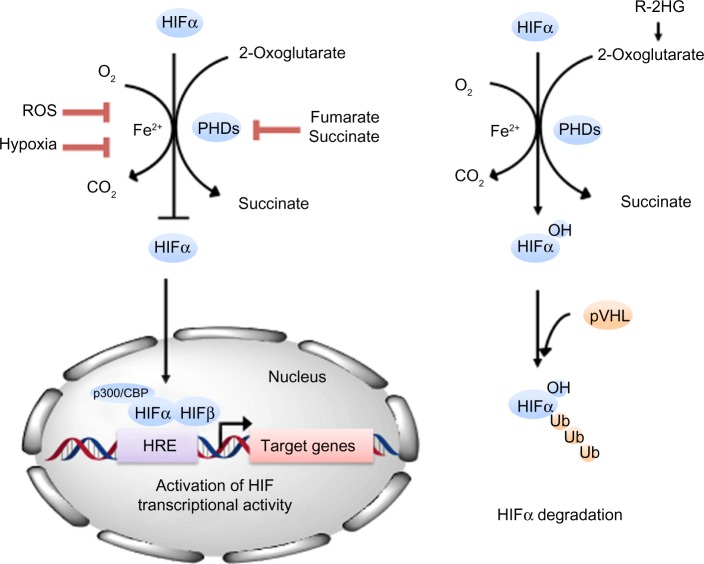
Three prolyl hydroxylases were identified in mammalian cells and shown to use O2 as a substrate to generate 4-hydroxyproline at residue 402 and/or 564 of HIF-1�� (2, 13, 24). These proteins are homologues of EGL-9, which was identified as the HIF-1�� prolyl hydroxylase in C. elegans by genetic studies (13). Alternative designations for the three mammalian homologues include EGL-9 homologue (EGLN), prolyl hydroxylase domain protein (PHD), and HIF-1�� prolyl hydroxylase (HPH) 1�C3. The hydroxylation reaction also requires 2-oxoglutarate (��-ketoglutarate) as a substrate and generates succinate as a side product. Ascorbate is required as a cofactor. The prolyl hydroxylase catalytic site contains an Fe(II) ion that is coordinated by two histidine and one aspartate residue. Unlike heme-containing proteins, the Fe(II) in 2-oxoglutarate-dependent oxygenases can be chelated or substituted by Co(II), rendering the enzyme inactive. Most importantly, these prolyl hydroxylases have a relatively high Km for O2 that is slightly above its atmospheric concentration, such that O2 is rate limiting for enzymatic activity under physiological conditions (13, 20).As a result, changes in the cellular O2 concentration are directly transduced into changes in the rate at which HIF-1�� is hydroxylated, ubiquitinated, and degraded. However, a thorough analysis of the relationship between O2 concentration and enzyme activity for each of the PHDs in living cells, and a comparison with the corresponding dose-response curve for HIF-1�� expression (29), has not yet been reported. In particular, the plot of HIF-1�� protein levels as a function of O2 concentration in HeLa cells yielded a sigmoidal curve suggestive of cooperativity (29), a finding that is not readily explained by the known biochemistry of the HIF-1�� prolyl hydroxylases.
Remarkably, HIF-1�� transactivation domain function is regulated by O2-dependent hydroxylation of Asn-803, which blocks the binding of the coactivators CBP and p300 (41). Factor inhibiting HIF-1 (FIH-1), which was identified in a yeast two-hybrid screen as a protein that interacts with and inhibits the activity of the HIF-1�� transactivation domain (44), functions as the asparaginyl hydroxylase (19, 40). As in the case of the prolyl hydroxylases, FIH-1 appears to use O2 and 2-oxoglutarate and contain Fe(II) in its active site (11, 42, 51), although it has a Km for O2 that is three times lower than the prolyl hydroxylases (37). Spectroscopic analyses of a peptide from the HIF-1�� transactivation domain complexed with the interacting domain of CBP or p300 revealed that Asn-803 is present within an ��-helix that is buried deep within the protein interface, where it participates in multiple hydrogen-bonding interactions that are predicted to stabilize the complex (9, 12, 15). Hydroxylation of Asn-803 is predicted to disrupt these protein-protein interactions.Similarly, hydroxylation of Pro-564 has been shown to also function as a molecular switch to positively regulate the interaction of HIF-1�� and VHL (21, 53). Thus hydroxylation provides a mechanism for regulating protein-protein interactions, similar to the effect of phosphorylation and other posttranslational modifications. However, what sets hydroxylation apart is that the modification occurs in an O2-dependent manner, thus establishing a direct link between cellular oxygenation and HIF-1 activity.
One remarkable aspect of the O2-sensing system described above is its plasticity. Although O2 may be the limiting substrate for hydroxylation under physiological conditions, it appears that under pathophysiological conditions iron or ascorbate may also be limiting (36). Furthermore, the expression of the PHDs varies from one cell type to another as well as in response to various physiological stimuli, including hypoxia (1, 10, 13, 52). Thus the O2dose-response curve may be shifted to the left or right under different developmental or physiological conditions. Alternative splicing of the primary RNA transcripts for two of the PHDs provides yet another mechanism for modulating prolyl hydroxylase activity (20). Finally, the transcriptional response elicited by a hypoxic stimulus also demonstrates a remarkable degree of plasticity, because the battery of target genes that is regulated by HIF-1 is unique to each cell type (34). Thus the identification of the molecular components of the O2-sensing system represents a milestone, rather than a finish line, on the course to defining the physiology of oxygen homeostasis.
Developmental and Physiological Consequences of HIF-1 Activity
The identification of HIF-1, VHL, FIH-1, and the PHDs over the past decade has delineated a pathway by which cells sense O2 and respond to changes in oxygenation with changes in gene expression, a property that is fundamental to the cells of all metazoan species. Coincident with these dramatic molecular discoveries have been equally dramatic discoveries regarding the remarkable variety of biological processes in which HIF-1 plays an important role. Analyses of mice, in which expression of HIF-1�� has been lost either in all cells (germline knockout) or a single cell lineage (conditional knockout), have identified multiple aspects of development and physiology that are dependent on HIF-1 (TABLE 1). Indeed, the study of HIF-1��s role in development and physiology provides a basis for unifying these two central areas of biology. O2 delivery to cells of the developing embryo becomes limited by diffusion such that establishment of a functioning circulatory system is required for embryonic survival by embryonic day 9 (E9) in the mouse. In wild-type mouse embryos, HIF-1�� expression increases dramatically between E8.5 and E9.5, whereas embryos that lack HIF-1�� expression die between E9.5 and E10.5 and show cardiac malformations,vascular regression, and massive cell death (7, 26, 39, 60). Complete HIF-2�� deficiency is also associated with embryonic lethality (58, 76), and because the embryos survive longer than Hif1a−/− mice, effects on multiple organ systems can be demonstrated (63).
TABLE 1. Developmental and physiological roles of HIF-1 as established by analysis of HIF-1��-null mice and cell lines Enlarge table
Whereas complete HIF-1�� deficiency results in developmental defects, partial HIF-1�� deficiency is sufficient to result in impaired responses to physiological stimuli. A particularly dramatic example is the loss of O2 sensing in the carotid body of Hif1a+/− mice (35). Although the carotid bodies are anatomically and histologically normal and depolarize normally in response to cyanide application, they show essentially no response to hypoxia. Thus partial HIF-1�� deficiency in the carotid body results in a complete loss of the ability to sense and/or respond to changes in the arterial Po2 by stimulation of the central nervous system cardiorespiratory centers. HIF-2�� is also expressed in the mouse carotid body (76), which suggests that HIF-1�� and HIF-2�� play distinct roles in this organ. The HIF-1 target genes that are critical for O2 sensing and/or efferent responses by the carotid body have not been identified. Remarkably, in the intact animal, other chemoreceptors are less sensitive to Hif1a gene dosage and compensate for the loss of carotid body activity in Hif1a+/− mice (35).
Another dramatic phenotype is the complete inability of Hif1a−/− myeloid cells (granulocytes and macrophages) to respond to inflammatory stimuli (8). Myeloid cells are dependent on glycolysis for ATP generation, perhaps reflecting the hypoxic microenvironment that is often associated with inflammation and infection. HIF-1�� deficiency results in ATP deficiency, which impairs critical myeloid cell functions such as aggregation, motility, invasion, and bacterial killing. The role of HIF-1 in immunity is not restricted to myeloid cells, because HIF-1 also plays critical roles in B lymphocyte development (38) and T lymphocyte activation (47). The ability to create mice in which HIF-1�� deficiency is restricted to a limited number of cell types (8, 62, 64) is likely to result in the identification of an increasing number of developmental and physiological processes that are regulated by HIF-1.
Medical Consequences of HIF-1 Activity The preceding sections provide a brief summary of the critical role of HIF-1 in understanding oxygen sensing, development, and physiology. HIF-1 plays an equally important role in disease pathophysiology, including ischemic cardio-vascular disease (3, 67) and cancer (68, 82), the most common causes of mortality in the US population. As a result, there is considerable interest in HIF-1 as a therapeutic target in these disorders (16, 68, 82). In the case of cardiovascular disease, increased HIF-1 activity induced as a result of HIF-1�� gene therapy (34, 72, 78), small molecule inhibitors of prolyl hydroxylase activity (20, 24, 43), or inhibitors of HIF-1��-VHL interaction (83) may provide a means to stimulate neovascularization of ischemic tissue. In contrast, small-molecule inhibitors of HIF-1 activity may be useful as anticancer agents (84). However, because HIF-1 functions as a global regulator of oxygen homeostasis, it may not be a useful therapeutic target if the treatment results in unintended and undesirable side effects. An alternative approach may be to focus on the products of HIF-1 target genes. For example, erythropoietin administration may reduce ischemia-induced apoptosis in patients presenting with acute cerebral or myocardial infarction (4, 14, 54, 57). The translation of a rapidly growing body of basic science data into clinical applications looms as the most challenging and most important goal in this exciting field.
AUTHOR NOTES
gsemenza@jhmi.edu
��
��
Hydroxylation of HIF-1: Oxygen Sensing at the Molecular Level | Physiology
https://www.physiology.org/doi/full/10.1152/physiol.00001.2004
��
Prolyl hydroxylase domain enzymes: important regulators of cancer metabolism
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5045062/
Hypoxia (Auckl). 2014; 2: 127�C142. Published online 2014 Aug
��

ͼһ��˵���������ǰ�֢�Ĺؼ����ء�DC,��ͻϸ��,EC,��Ƥϸ��;GLUT,������ת����;IL,����;HAT,�鵰��������ת��ø;LDH,��������ø;MCT,������ת����;PEP,����ϩ����ͪ��;PGAM,�������������λø;PPP����������;��;��������������;TAF,������س���άϸ����
Lactate: A Metabolic Key Player in Cancer | Cancer Research
https://cancerres.aacrjournals.org/content/71/22/6921.long
��
��
��
��
��
NDRG3��������ΰ�����Ԥ�����й�
NDRG3 overexpression is associated with a poor prognosis in patients with hepatocellular carcinoma
��
�о����������մ�ѧ������������ҽԺ
ȱ���յ�����(HIFs)��N-myc���ε��ػ���3 (NDRG3)�ı����ܸ������ǻ�ø��(PHD)ø���������Ե��ء�
N-myc���ε��ػ�����һ���·��ֵĻ�����壬��NDRG1��2��3��4��ɡ�NDRG���ͨ�����ذл���ת¼��Ӱ��ϸ����ֳ���������ֻ��ͷ���[23,24]����ˣ�����������������������������������Ҫ������[25,26]��ϵͳ������������������NDRG1��NDRG3����һ���Ǽ��壬��NDRG2��NDRG4������һ��[10]�Ǽ��塣
N-myc���ε��ػ���3
(NDRG3)��NDRG�������Ҫ��Ա������ϸ����ֳ���ֻ�������ѧ���̡����о�������NDRG3�ڸ�ϸ����(HCC)�еı��̽����NDRG3��HCC�����еı��������ٴ����������Ĺ�ϵ�����Ƕ�HCC��֯����ʵʱ������ת¼�ۺ�ø����Ӧ(qRT-PCR)�������黯(IHC)�������Բ���NDRG3��HCC�����еı�������������Kaplan-Meier�������ߺ�Cox�ع��������102��HCC���ߵ�Ԥ�����ʾ�����������֯��ȣ�HCC��֯NDRG3�����������ߡ����⣬���ǵķ�����ʾNDRG3������������С(P=0.048)�Ͳ����ּ�(P=0.001)��ͳ��ѧ���塣���������Kaplan-Meier������ʾ��NDRG3������HCC����������(P=0.002)��������(P=0.005)�Ķ���Ԥ��ָ�ꡣ��Щ���ݱ�����NDRG3�ı�����ܱ���Ϊ��һ���°��������־���Ԥ��HCCԤ����·�����
��
����
��ϸ����(HCC)������������Ķ�������֮һ���ڶ��������У�HCC�Ļ�����ȫ���������壬������ȫ�������ڶ�����2015�꣬����20,000��HCC������������������[2]�����й���HCC�·���������������ռȫ���ܲ���(40����)��һ�����ϡ��й���������ʡ�������������ϸΰ���������ߵĵ�����HCC�ķ�����չ��һ�������ء���Ρ������Ĺ��̣��������ס������ײ�����Ⱦ���ƾ����ˡ��Ǿƾ���֬���ε��������[5,6]����20�������ΰ������绯�ơ������ڡ������г�����Ƶ������ֲ[7]�Ⱦ��н�չ�����ΰ��ĸ�ת�ƺ߸�����˵��������Ԥ���Բ����롣���ǵ���һ�㣬�Լ�Ԥ��������صĸ����ԣ�����ȷ��Ԥ��Ԥ�������ԸĽ�HCC���ٴ����ơ�
N-myc���ε��ػ���3
(NDRG3)��NDRG�������Ҫ��Ա֮һ����ϸ����ֳ���ֻ�������ѧ�����з�����Ҫ����[9,10]��NDRG3��غ�衢�ѳ���ǰ���١������ԭʼ�����и߱����ת¼�����ڴ����б�����ߣ���������������[11]��NDRG3�������侫����Ƥ����㣬��ʾ����ܲ����˾����γ�[12]�Ĺ��̡���������NDRG3�����������еĸ����������������ǵĹ�ע��NDRG3�ٽ�ǰ���ٰ���֯��ϸ��������ϸ����NDRG3�Ĺ��ȱ�����ϵ�CXCL1��CXCL3��CXCL5��Ѫ���������ӣ�����Ҫ�����Ǵٽ�Ѫ���������ٽ���������[13,14]��NDRG3�������ײ���(HBV)��صĸ�ϸ�����й������ˣ�����HCC[15]��һ��DZ�����ưе㡣NDRG3������Ҳ����Ϊ���Сϸ���ΰ�(non-small
cell lung cancer,
NSCLC)��ǰ���ٰ��ͺ����۰����[16-18]����Щ�����ʾNDRG3���д���������;Ȼ���������һ�ݱ��������NDRG3�µ����ܲ������ٰ��ķ����ͽ�չ������[19]��Ȼ����Ŀǰ��û���о�̽��NDRG3��HCC�е�Ԥ���ֵ������Ҫ��һ���IJ�����
�ڱ��о��У�����ʹ��ʵʱ������ת¼�ۺ�ø����Ӧ(qRT-PCR)������NDRG3��HCC�걾�����ڽ���������֯�еı�����⣬���ǻ���������֯оƬ(TMA)�������黯(IHC)�����NDRG3��HCC��֯�еı��������ǻ�������NDRG3������HCC�����ٴ����������Ĺ�ϵ���ر���NDRG3������Ԥ�������Ĺ�ϵ��
����
N-myc���ε��ػ�����һ���·��ֵĻ�����壬��NDRG1��2��3��4��ɡ�NDRG���ͨ�����ذл���ת¼��Ӱ��ϸ����ֳ���������ֻ��ͷ���[23,24]����ˣ�����������������������������������Ҫ������[25,26]��ϵͳ������������������NDRG1��NDRG3����һ���Ǽ��壬��NDRG2��NDRG4������һ��[10]�Ǽ��塣Ŀǰ������NDRG1�������еı�����ڲ�ͬ�Ĺ۵㡣һ���о�������NDRG1����ͨ�����������������յ�ϸ����������Ϊ��ֱ����(CRC)���ְ����ӡ�������˵��NDRG1�������march
-8�յ�������4�Ľ��⣬����NDRG1��CRCϸ�����������������Լ������У�����������������صĵ����յ�����(TRAIL)[27]��NDRG1��ʳ���۰�(ESCC)�еĹ�����Ҳ����������Щ���ߵĶ�OS��ء��ݱ�����NDRG1��ESCCϸ���й�����ɼ���Wnt�ź�ͨ·���յ���Ƥ-����ת��(EMT)���Ӷ�����E-cadherin�ı��������ţ�ı���[28]��������������NDRG2����������E3��������øSkp2���ԣ��յ�CRCϸ���ֻ�Ϊ[29]��Hypoexpression
NDRG2Ҳ���ܼ����յ�EMT
NF-��B�ź�ͨ·,�Ӷ�������ӵ������ʹ�С��ǻ��״ϸ����(OSCCs)���ֵĿ������Լ������ܰͽ�[30]������NDRG3��NDRG4�о�������NDRG4�ڽ�ֱ�����ͽ���ĸϸ�����о����ְ�����[31-33]��NDRG3�ڼ��ְ�֢�ж��б������ݱ��������״���ģ�������תȾ��PC-3ϸ����ȣ���Դ��NDRG3�����������ɵ��¿�¡������Ǩ�������������ٶ����ӣ����������ڹ�����NDRG3�����������ֲ������������Щ�����ʾNDRG3��ǰ���ٰ��ķ����ͷ�չ����ؼ����á�Fan����[15]������NDRG3��HCC�걾�б����ϵ�������NDRG3�ɽ��ΰ�ϸ���Ķ��Ա��͡������һ���о�������NDRG3�ϵ���NSCLC�걾��ϸ��ϵ�о��ɼ�;��ˣ�����������NSCLC[18]��һ���µ�Ԥ�����ӡ�NDRG3���о���Ҫ������ȷ��������ѧ���ơ��ݱ�����ͨ��NDRG3-Raf-ERK��������յ���Ӧ������ά�������ڳ���ȱ�������µĽ�չ[34-36]����֤�ݱ���NDRG3��һ���µ����õ����֢�����־��;Ȼ���������б�����NDRG3��HCC�걾�б����ϵ��������ٴ��������弰����HCCԤ��Ĺ�ϵ�в������������Ҫ������о���ȷ��NDRG3��ΪHCC��DZ�����ưе㡣
���ǵ�qRT-PCR�����ʾ��HCC�걾��NDRG3
mRNAˮƽ����������������֯��ͬ����TMA��IHC����Ҳ��ʾ��HCC��֯�е�NDRG3����ˮƽ�������ڷǰ���֯����Щ������������о����һ��[15-18]����ͬ����NDRG3�ڶ��������б����ϵ������⣬NDRG3��HCC�걾�е����Ա�����ijЩ�ٴ���������(��������С�Ͳ����ּ�)������ء�Du����[37]���miR-31���ϵ�����ͨ������NDRG3�ı���������HCC�ķ�������������֮ǰ���о����һ�¡���Щ�����һ������NDRG3������HCC��������ؼ����á�
�����غͶ����ط�����ʾ��NDRG3������HCC���ߵ�DFS��أ�������ΪDFS�Ķ���Ԥ�����ӣ���NDRG3�����������С����Ϊ��Ӱ��OS�Ķ���Ԥ�����ء����⣬Kaplan-Meier������������NDRG3�������ԵĻ��߱�NDRG3�������ԵĻ����������̡����������о����һ�£�NDRG3�������еĹ�����ٽ��������ķ�չ�����벻��Ԥ�����[16-18]��
�෴�����о�����NDRG3��ijЩ�������������ٰ��б��オ�ͣ��Ӷ����ֳ��ְ����á����ֲ������������������Դ��DZ�ܵIJ�ͬ
NDRG3�ڲ�ͬ���������еĹ��ܲ��죬��NDRGs[38-40]��
���о�����һ���ľ����ԡ����磬����û�м�NDRG3���ܰͽ�����ϸ���еı���������NDRG3����HCC������ת�ƵĹؼ������⣬���о���TMA�걾Ϊ��������TNM�ڡ�HCV��Ⱦ��HCC��������ʷ�˽�������ȱ����Ϣ;��ˣ���Щ�����������ƫ���ġ����ǽ������Ľ����ǵ�ʵ����ơ��������������о������NDRG3��HCC�걾�еı�����죬���״�̽����NDRG3������HCC�����ٴ������Ĺ�ϵ���ر���NDRG3��Ԥ���ܡ�NDRG3����Ϊ��HCC�����µ�Ԥ���־�����ΪHCC�����ṩ�µ����Ʒ�����
��
NDRG3 overexpression is associated with a poor prognosis in patients with
hepatocellular carcinoma | Bioscience Reports | Portland Press
https://portlandpress.com/bioscirep/article/38/6/BSR20180907/98276/NDRG3-overexpression-is-associated-with-a-poor
��
NDRG3��NDRG4�������µ�������ػ���
N-myc���ε��ػ���NDRG3��NDRG4����Ϊ������ѧ���̺ͷ��������з�����Ҫ���á�NDRG3��NDRG4�ı�������������ϸ��ϵ�����������б����ּ��ٻ�ȱʧ����ʾ���ǿ��ܷ����ְ�����Ĺ��ܡ�����������NDRG3��NDRG4�ķ��ӽṹ��ϸ������֯�ֲ�������ѧ�����Լ��������е����õȷ�����о���չ��������ͼչʾ�����ڼ����о��е���Ҫ�Ժ�����DZ����
�����
a.����ҽ�ƴ�ѧ��ɽ������
b ����ҽ�ƴ�ѧ����ҽԺ��ɽ������
c �й�ɽ��ʡ����������������ҽԺ
NDRG3 and NDRG4, two novel tumor-related genes - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0753332213000632
��
��
��������Ķ�ȱ���ķ�Ӧ
A Lactate-Induced Response to Hypoxia
Medical Genomics Research Center, Korea Research Institute of Bioscience and
Biotechnology (KRIBB), Daejeon 305-806, Korea
ͻ����
NDRG3��PHD2/VHLͨ·�������ڵ���
•������NDRG3��ϣ���������ȱ��״̬�µ�ˮƽ
NDRG3����rafe - erk�źţ��鵼����������ȱ����Ӧ
�ܽ�
����������ܹ�����������ƽ��Ͳ��������¶Ե���������Ӧ��ͨ�������յ�����(HIF)���ڵ�����Ӧ�Ѿ��õ��˺ܺõ�֤ʵ��������֤�ݱ�����������HIF�صĻ���Ҳ�������С���������DZ�����һ��������������۵�ȱ����Ӧ��������һ�ִ�л��������ȱ�����������ӡ����Ƿ���NDRG3�����ڳ�����������PHD2/
vhl�����ķ�ʽ���⣬��ͨ������ȱ�������»��۵������϶������ƻ����ȶ���NDRG3�����c-Raf�鵼ȱ���յ���raferkͨ·����ٽ�Ѫ�����ɺ�ϸ������������ϸ��������IJ���������ndrg3�鵼��ȱ����Ӧ����ˣ����ǵ��о������������յ�ȱ���źŴ����ķ��ӻ�����Ϊ�������ȱ���յ����������Ʒ����ṩ�����ݡ�
��
����
����̬�Ǻ�����������ѧ�Ļ������ڵ��������£�ϸ������ȱ���յ��ķ�Ӧ����Ӧ�������ڶ��ӵĻ���(Cassavaugh��Lounsbury,
2011)��ȱ����Ӧ����̥�����ͳ����������������ܵ���Ҫ��ɲ��֡�����Ҳ�����༲���IJ��������ɷ֣�������֢����֢����Ѫ�ܼ�����
�����յ�����(HIFs)ͨ������һϵ�е�����Ӧ����ı����ڲ�ͬ�Ĺ����з������ã������³´�л�������ݡ�pH���ڡ�Ѫ�����ɡ�ϸ����ֳ�����棬�Ӷ��ڵ�����Ӧ�з��Ӻ�������(Harris,
2002, Cassavaugh��Lounsbury,
2011)���ر��ǣ�hif�鵼���ǽͽ��ϵ�������������(TCA)ѭ����ȱ�����ڵ�һ���ؼ�����Ӧ�Է�Ӧ(Cassavaugh��Lounsbury,
2011)�������յ����ӵı���ͻ�����������Ե��ǻ����ϸ��ܵĦ��ǻ�(����,2003)��
Խ��Խ���֤�ݱ�����ȱ��������治�ܵ�����hif�鵼�Ļ��������͡����磬����hif�鵼��ͨ·����������ֹ��������;������Դ��HIF-1��-deficient��̥��ϸ��(ES)ϸ�������������ڵ����յ�ϸ�������ļ��ٺ�����ѹ���յ���ֳ(Carmeliet
et al .,
1998)�����౨������������Ѫ��������hif��������������Ҫ;������ˣ���HIF1A��ESϸ�����ó�ʱ��Ѫ�����ɵ��Ա���(Hopfl�ȣ�2002)��һЩ֤�ݱ�������Ѫ���������ӣ�Ѫ����Ƥ��������(VEGF)����ͨ��hif������hif����;���յ�(Mizukami
et al.�� 2004)���յ�����pro-angiogenic��������������HIF-1��-deficient�᳦��ϸ����Ѫ�����ɷ�Ӧ(Mizukami et
al .,
2005)�����⣬��HIFs�⣬��֪�ж���ͨ·��ת¼����(TFs)��ȱ��������Ӧ����һ����hif�صķ�ʽ�յ����ﷴӦ������Щoxygen-regulatable
TFs
NF-��B��AP-1��CEBP������ȱ��(����˹��̩��,2005)����ˣ��м�ƪ����������ȱ�����ص�һЩ������HIFs���أ���ʾ����������ͨ·������������HIFͨ·����prolyl�ǻ�ø��(PHD)ø����(Elvidge
et al.�� 2006)�����⣬���൰��ø��PKA��PKC��PI3K��AKT��JNK��PTK2B (Pyk2)��SRC��MAPK14
(p38)��ERK1/2��ȱ�������±�����(Seta et al.��
2002)��Ȼ�������ܽ�����������Щ�о���ȱ����Ӧ����hif�صķ�֧�����������ڵĹؼ����غͻ�����Ȼ�������
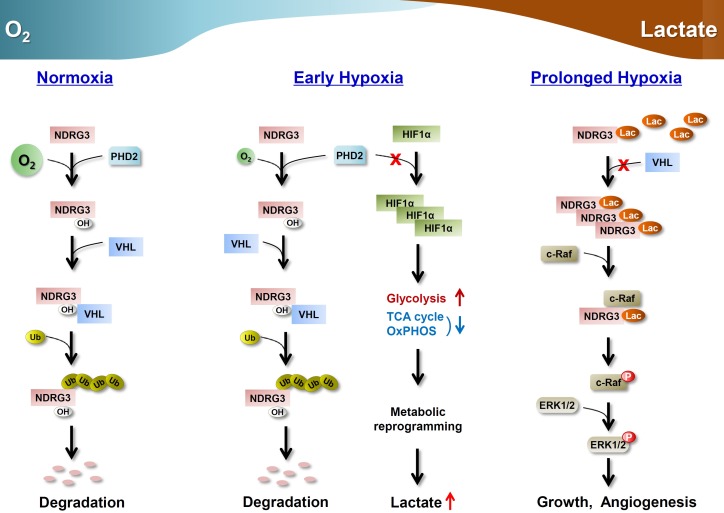
�ڱ��о��У����Ǽ�����һ�������ڵ��ף�NDRG3
(NDRG�����Ա3;NM_032013)����ΪPHD2/VHLϵͳ����ʵ���ʡ�NDRG3�ڲ�ͬ��ϸ�������У���ȱ�������¾��иߵ��յ����ԣ�����mRNA�ı��ﲻ��HIFˮƽ��Ӱ�졣��Ȥ����,�ǽͽ�NDRG3��Ҫ�����ղ�ƷΪ��ȱ����������,����������������ڵ����յ����ӱ���HIF-1�����ڵ�����������ø�ı���(LDHA)�����Ƿ���NDRG3�������յ���ȱ���ź�����ؼ����ã���ͨ���鵼raferkͨ·�ļ������ٽ���ʱ��ȱ��ʱ��Ѫ�����ɺ�ϸ����������ˣ�NDRG3Ϊ����ȱ����Ӧ�������������������ṩ��һ���ؼ����Ŵ����ء�
���
����NDRG3ΪPHD2�ĵ���
Ϊ��ʶ��ȱ����Ӧ�ĵ������ӣ�������MCF-7ϸ����ͨ�����ΪPHD2�����߳������������÷�Ѱ��PHD2��ϵ��ס���ѡ���зḻ�ĵ������ֶӿ���������չʾ�����߳�����Ӧģʽģ���PHD2-Flag����֮��,����ѡ��NDRG3���н�һ�����о�,��Ϊ�����ڻ��������ϸ����ֳ��Ǩ�ơ������Լ��ڷֻ��ͷ�չ(Melotte
et al ., 2010),������ȱ�����﹦���������(2011��,2002��Ĺ���˹Cassavaugh���Ͷ�˹����)(ͼS1A)��
Ϊ����ϸ����NDRG3��������NDRG�����Ա�п�����һ�����NDRG3���ʹ������¡����(ͼS1B)���ÿ�����PHD2-Flag���߳���Ƭ���м�NDRG3Ϊ42-KDa����(ͼ1A)������ͨ�����߳�����Դ��NDRG3��ȱ��������HeLaϸ���е�PHD2-Flag����֤NDRG3-
phd2�������(ͼ1B)����ֱ��ʹ������PHD2-His��NDRG3-
gst����������ʵ��(ͼS1C)����ˣ�������ΪNDRG3��һ��������phd2��ϵ��ס�
��
Ȼ������ʹ��PHD���Ƽ�desferrioxamine
(DFX)���PHD2��NDRG3֮����ܵĹ��ܹ�ϵ����ȻNDRG3�Ļ���ˮƽ������Ժ��Բ��ƣ���PHD���Ƶ�������HeLa(ͼ1C)��MCF-7ϸ���еļ��������Ի���(ͼS1D)����Щ�������������PHD���Ƽ�������xaloylglycine
(DMOG)��CoCl2(ͼS1E)�ظ�������NDRG3���ױ�������ܵ�PHD�鵼�ķ������ơ�Ȼ������ʹ��С����rna
(sirna)������˲�ͬ��PHD�����Ա�ڳ���������ͨ����Ĭ�����������NDRG3�ĵ��ء������������,����HIF-1��,PHD2
NDRG3�������Ҫ��ܻ����IJ�ʿ��ͥ��Ա(ͼ1
d,��)���ڹ����߳��������У�NDRG3��PHD2֮��IJ�������õõ���֤ʵ(ͼS1F)��VHL��ȱʧ��E3��������ø������İ���Ԫ����Ҳ������NDRG3�ڳ��������µĻ���(ͼ1D����)����ʾNDRG3������PHD2/VHL�鵼�ķ�������εİе㡣Ϊ�˸����ؽ����һ�㣬�������˼���NDRG3���ı��壬����Я����һ�����ᣬ�ڼٶ���PHD2-�Խ�λ���Ϸ����仯������ͨ���ٶ���NDRG3�ṹ���ѷ�����PHD2�ṹ֮��ĶԽ�ģ��Ԥ���(Chowdhury
et al.�� 2009)(ͼS1G)�������߳���ʵ�������NDRG3ͻ������Ը�����phd2���ǿ����������:V296D > Q97E >
R47D��N66D����Ȥ���ǣ����ƺ��������ڳ��������µĵ��ױ���ˮƽ�ʸ����(ͼ1E)�����⣬NDRG3ͻ�����PHD2�����߹�������ha��ǵ�VHL����������(ͼ1E)������NDRG3��PHD2��VHL����������䵰�ױ���Ĺؼ��������ء��������������ڷ��ػ�ʵ���У�NDRG3�����������NDRG3���߳����ķ�����������ͬ�Ķ̷���rna
(shRNAs)��Ĭ�����ɽ��ͷ�����(ͼ1F��S1H)�����⣬MG132���Ƶ���ø�����������HeLaϸ����NDRG3�ļ��ˮƽ(ͼS1I)������������NDRG3��һ����PHD2����õĵ��ף��������PHD2/
vhl�鵼�ĵ���ø��;�������ء�
����������NDRG3���ı���
����PHD2�Ļ����ںܴ�̶���������O2����Ч�ԣ����Ǽ����NDRG3���ı����Ƿ����������ĵ��ء�NDRG3��MCF-7ϸ���еĻ���������O2Ũ�ȳʸ����(ͼ2A��S2A)�����һ�£�ȱ��������HeLaϸ��NDRG3���ػ�����������(ͼ2B)��NDRG3�ĵ����յ��ڲ�ͬ��֯��Դ�İ�ϸ���ͷ�ת��ϸ���ж��õ���֤ʵ(ͼS2B)�����������һ������ձ��ԡ�Ȼ��,���֮��HIF-1����������ʾһ�����θ�Ӧģʽ��ȱ�������ڽ�,NDRG3չ��һ��s�εı���ģʽ,��ʼ��HIF-1��ˮƽ��ʼ�½�,��һֱ����������ȱ��(ͼ2)������ϸ��������NDRG3�ĵ�����������(ͼS2C)����Щ���ǿ����ʾNDRG3���ױ������������ء�
�������������о���NDRG3���ױ�����������Ե��صķ��ӻ�����������������NDRG3�ڸ�����294���������ǻ�����������PHD2���κ�IJ���(ͼ2D)������ͻ��ĸ�����294��������(P294A)�������Եı��쵰�Ļ����ڳ���������(ͼ2E����)�����⣬�����߳���ʵ���������Ұ������ȣ�P294Aͻ�䵰��PHD2��VHL���Ľ���������Խ���(ͼ2E����)�������������294��PHD2�鵼���ǻ����Ĺؼ���λ�㣬��������NDRG3�����ڳ��������µ��ȶ��ԡ�
��HIF-1����������֮ǰ��NDRG3(ͼ2 c),���ǵ���Ŀ�����HIF-1��ת¼����NDRG3����ʽ��ȱ����RT-PCR������ʾ��ȱ��ʱNDRG3
mRNAˮƽ�������ֲ��䣬��ʹHIF���״ﵽ��ֵ(ͼS2D)����һ���������NDRG3ת¼��HIF�����ԣ���֤ʵ��NDRG3��ȱ�������±���ķ�������ʡ�HIF��ͬ�ǻ���ȱʧ��NDRG3
mRNAˮƽ��Ӱ�죬֤ʵ����ת¼��HIF������(ͼ2F)��ֵ��ע����Ǿ���NDRG3���ױ�����HIF-silencedϸ��ȱ����Ȼ�ɼ��,��������������Ϳ���HIF-1�»���,��һ��С�ö�ij̶���,��HIF-1������,��ʾDZ��non-transcriptional
HIFͨ·NDRG3�����ʱ����Ӱ�졣ͬʱ�����ǿ��Է���HIF����һ��NDRG�����ԱNDRG1���������ת¼��������(ͼS2E)����Щ�����ͬ������ȱ��ʱNDRG3�����ת¼���ز�����ҪHIF���ԡ�
NDRG3�ڵ���ȱ����Ӧ�е�����
����ͨ����NDRG3�ĵ��ױ����������ȱ����Ӧ������������۷���Ļ���������������о���NDRG3��ȱ���е�DZ�ڹ���(ͼS3A)��ͨ���������������ƻ������۵Ļ�����������ȱ��ʱ�ض�ʱ���Huh-7ϸ��ת¼��������ݼ�������������(Z����)�����������NDRG3���ױ����롰Ѫ�����ɡ�������������������ֳ(����)�������˶������ܸ߶���أ��롰�ǽͽ⡱��(ͼ3A)����һ���棬NDRG3��ȱ��24Сʱʱ�Ľߣ���ϸ����NDRG3���ױ��ﱾӦ�ﵽ����ˮƽʱ�����¡�Ѫ�����ɡ�������������������ֳ(����)�������˶�������Է��������仯�������ǽͽ⡱�����δ���������仯(ͼ3B)�����֮��,���ǽͽ⡱���Ե�Ŀ��HIF-1������6������Դ��ȱ��,��HIF-1�����ױ���Ԥ�ƴﵽ��ֵˮƽ(ͼS3B)��һ�µأ�NDRG3��һ�������ȶ��������λ����(ͼ1E�е�N66D)�����˾���Ѫ�����ɡ���ֳ�����������ֵ�������Ǩ�ơ��ǽͽ����Ҫ���ܵĻ�����ϵ�(>1.5��)(ͼS3C)��
��
Ȼ�����Ƕ�NDRG3�ڡ�Ѫ�����ɡ��������������͡���ֳ���е����ý�����ʵ����������Щ����ͨ���������������йأ�����NDRG3�ĺĽ���������(ͼ3B)����HUVECϸ���ɹ�ʵ���У�NDRG3�Ľ�����������ȱ���յ���Huh-7ϸ����Ѫ�����ɻ���(ͼS3D)�����ͬʱ������������ʵ�����NDRG3�ó�������BALB/c-nuС��Huh-7ϸ����Ѫ�����ɻ���(ͼ3C)���ڷ���ˮƽ�ϣ�ȱ���յ��Ĵ�Ѫ�����ɱ����ı��ﱻNDRG3�ľ�����NDRG3�ϵ�(N66D)(ͼ3D)����������ͨ��caspase-3/7��PARP�ѽ�ʵ����NDRG3�Ŀ��������ԣ�����NDRG3�ĺĽ������ٽ�ȱ��ʱ��ϸ������(ͼ3E)����ˣ�ȱ���յ��Ŀ���������ı���ر���IAP(�����������Ƽ�)����ij�Ա��ͨ��NDRG3�ĺĽ���Huh-7ϸ���б�����(ͼ3F)�����⣬ʹ�������3��-UTR��shRNA(ͼS1H�� #5)����NDRG3�������������ȱ��(3% O2)��Huh-7ϸ��������;�������ֱ��ͱ�ȱ��NDRG3��Ȼ3��-UTR���е�����NDRG3(N66D)����������Ч����(ͼ3G��S3E)�����⣬NDRG3�����ó�������������BALB/c-nuС��Huh-7ϸ������������(ͼ3H��S3F)����Ȥ���ǣ�NDRG3��HIFs�е��κ�һ��ͬʱȱʧ����ȫ�����������������������NDRG3��HIFs��ȱ��ϸ�������о��л�������(ͼ3H)���г�����������ӫ��������ʾ��NDRG3��ȱʧ��Ч������������Ѫ������(IL8��CD31)��ϸ����ֳ(Ki-67)��־��ı����hifȱʧ�����ı�־�����ˮƽ��������൱(ͼS3G)�����֮�£�NDRG3(N66D)����λ����������֬�и߶ȴٽ���Huh-1ϸ���ļ����γ�(ͼS4J)�Լ�BALB/c-nuС�����������(ͼ3I��S3H)����Щ�������NDRG3��ȱ��ʱ�ٽ�Ѫ�����ɡ���������ϸ����ֳ�������Źؼ����á�
��
l-���ᴥ��ndrg3�鵼��ȱ����Ӧ
HIF-1�����,��ʾ��ȱ�����ڸ�Ӧģʽ,Ѹ����ʧ�ڸ�����ϸ��,NDRG3��ʼ�������ȱ������ˮƽ�����½���������(ͼ2
c��S2C)��NDRG3�Ļ��ۺͽ�����ڽϳ����ͺ��ڣ���ʾ�������������漰�����ء���ˣ�����̽���˳�����ˮƽ���롰��ʱ��ȱ������ص���������������NDRG3���ױ�����ϸ�����������߶����;ȱ��ʱ��NDRG3���ı��↑ʼ��∼6Сʱ���������εIJ���ģʽ�dz��ӽ�(ͼ4A)����һ���棬ʹ��LDHA���Ƽ�-���������������εIJ����������Լ��������ķ�ʽ�����Ե�����NDRG3���Ļ���(ͼ4B)�����Ƶأ�ͨ��sirna�鵼��LDHA����(ͼ4C)��2-���������ǵ��ǽͽ��ж�(ͼS4A)�����������ε����ɣ���������ȱ����NDRG3���ı�������Ǻ�/��Ȱ��������ǽͽ�Ȱ������ֽ���������ֱ��ǵ���ϸ���ڲ��������������Ҫ��л;����ͨ������ϸ���������Ǻ�/��Ȱ�������NDRG3���Ļ���Ҳ��֮���٣�������Ӱ��NDRG3��ת¼(ͼ4D)��Ȼ�����������ǰ�������������ȣ��Ȱ�����ЧӦ�ƺ���Խ�С���෴���ٽ���������(ͨ��LDHA�������/���ͪ���ʳ;ͼS4B)������ڻ���(ͨ�����MCT4�ij���;ͼ4C)��ǿ��NDRG3����ȱ�����ۡ���Щ�����������HIF���ײ�ͬ��ȱ�������������Ե���NDRG3���Ļ��ۣ�������Ҫ�ǽͽ�������ᡣ
��
Ȼ������ͨ����ϸ���ṩ��Դ����������ֱ�ӵ���֤�����NDRG3������ѧ��Ӱ�죬��Щϸ����ϸ��������IJ����ܵ��Ŵ���ҩ���ֶε�Ӱ�졣�������Ӽ���������Huh-1ϸ���ָ�����ȱ��NDRG3���ױ���,����LDHA��Ĭ,��Ӱ��NDRG3
mRNAˮƽ��HIF-1������(ͼ4
e)���������ǰ���(ͼS4C)������δ���(ͼS4D)��������IJ���ʱ��Ҳ�õ������ƵĽ����Ȼ������MCT1Ϊ�е��siRNA�ƻ�������鵼��NDRG3���ױ���ָ�(ͼ4F)�����ǻ��۲쵽���ڹȰ����δ����������ǰ����Huh-1ϸ���У�MCT1���µ�Ҳ�����Ƶ�Ч��(ͼS4E��S4F)��������������Щ�������NDRG3������ȱ��״̬����Ҫ������ۣ���˵��NDRG3������Ϊһ��ȱ���յ������ᴫ����������ϸ����hif�ص����ﷴӦ��
��ˣ������о���ȱ��NDRG3�����������л�еĹ������塣�����ȱ�������£��ò�������������IJ���������Huh-1ϸ�������ļ�������������(ͼS4G)��Ȼ����ͨ���������NDRG3(N66D)��Ч���������һЧӦ(ͼS4G)������NDRG3�����������յ���ȱ��ϸ�������з��ӹؼ����á���Ȼ��NDRG3(N66D)���ܹȰ����δ�����ֱ��Ӱ��(ͼS4H)�������Ҳ���ܹȰ����ε�Ӱ��(ͼS4I)��˵��NDRG3(N66D)����ȷʵ�����������á�ʹ��Huh-1ϸ����Ⱥ���γ�ʵ���һ��֤ʵ�˹Ȱ����ζ�NDRG3(N66D)ϸ�������������ȵ�Ӱ��(ͼS4J)��Ȼ�����Ǽ����NDRG3��ϸ�������е����ã���Щϸ����LDHA���ﱻRNAiȥ����shRNA��LDHA������������Huh-1ϸ�������ȱ�������µ�����(ͼS4K)�Լ�BALB/c-nuС�����������(ͼ4G��S4L)��NDRG3(N66D)�������ⶼ��Ч���ֲ���LDHAȱ�ݡ����⣬��HUVECϸ���ɹ�ʵ���У���ȱ�������£�oxamate������Huh-1ϸ���յ���Ѫ�����ɻ���(ͼ4H)��Ȼ����NDRG3(N66D)����λ����ָ�����Щϸ����Ѫ�����ɻ��ԣ�������oxamate����ˣ������ƺ���ȱ��ϸ��������Ѫ�����ɵĹؼ��źţ���NDRG3�������յ�ȱ����Ӧ�Ĺؼ��н顣
�������NDRG3���ױ���ķ��ӻ���
ͨ���о������NDRG3���ػ���Ӱ�죬̽���������յ�NDRG3�����۵ķ��ӻ��ơ������⣬������ͨ��HEK293Tϸ����PHD2/VHL���������߳�����NDRG3���ػ�(ͼ5A)�����������ο������PHD2/VHL��NDRG3�������Ρ�������Ȼ����Ӱ��HIF-1�����ױ�����ȱ��(ͼ4��ͼS4)����ˣ�����ͨ���о������ַ���֮�����������о�������ֱ�ӵ���NDRG3�Ŀ����ԡ�����gst��ǵ�����NDRG3����[14C]��ǵ�l-�����ν��е�������ʵ�������NDRG3�������Ϻ�ֱ�ӽ��������(ͼ5B��S5B)��Ϊ�˽�һ����֤NDRG3-��������ã�����ͨ���Խ�ģ��(δ��ʾ)Ԥ����NDRG3�ٶ�����������Ԥ�����������ͻ��������䲿�ְ�����л���ͻ�����ͻ�䵰�ĵ�������(ͼS5A)������һ��glycine-138ͻ��Ϊɫ����(ͼS5A�е�N3(G138W))��ͻ������ȱ�������¼���û�л��ۣ�����MG132���ڵ�����»��ۣ������������ʧȥ���ӱ�PHD2/
vhl�鵼�ĵ���ø�彵�����������������������ʵ�ϣ����ǹ۲쵽����N3(G138W)-GST������������ʵ���������ص���������������(ͼ5C��S5B)����Щ������������������Ƶ���ø�彵���NDRG3���������PHD2/VHL��
���⣬NDRG3-���Ḵ����һ���γɣ��ƺ���Ȼ��PHD2/
vhl�鵼�����ξ����൱�Ŀ��ԣ���Ϊȱ�����۵�NDRG3�����ڳ��������µ�������������������ά����һ��ʱ��(ͼS2C)�����֮��,HIF-1��Ѹ����ʧ�ڸ���,չʾ�������������Եķ����ļ�ܡ�
����ͨ����HEK293Tϸ���б����λ��ǵ�NDRG3��PHD2��VHL�ĵ���Ϸ�������һ��̽���������յ�NDRG3����̬�仯�Ļ��ơ�NDRG3��PHD2��ȱ������(6Сʱ)��ȱ������(24Сʱ)�Ľ���볣��״̬�����������죬��ʾ�����������ˮƽ����Ӱ��NDRG3-PHD2�������(ͼ5D)�����֮�£�NDRG3��VHL��ȱ��24Сʱʱ������Լ��٣�����ȱ��6Сʱʱ��ά���ڳ���ˮƽ����˵��������ˮƽ���ǵ���ˮƽ����Ӱ��NDRG3-VHL������á�Ȼ������ʹ����PHD2
(P294A)��������(G138W)�о���prolyl�ǻ���ȱ�ݵ�NDRG3��������֤��Щ�۲�����Ұ���ͻ����NDRG3�����ڳ�����ȱ��(24Сʱ)�����µ�phd2�������������������(ͼ5E)����һ���棬Ұ����NDRG3��vhl���������ȱ�������½ϳ������������Խ��ͣ���P294A��G138W��vhl���������ȱ�������¼���û�б仯��ֵ��ע����ǣ�P294A��VHL������ÿ��Ժ��Բ��ƣ���G138W-VHL����������ڲ�����������������µõ���ǿ�ҵ�ά�֡�Ұ����NDRG3�ķ��ػ���ȱ���������������ͣ���P294A��G138W�ڳ�����ȱ�������µķ��ػ����ɺ�����ǿ�Ƚ�ǿ(ͼ5F)���෴��oxamate���������Ե���ǿ��Ұ����NDRG3��VHL�ĵ���������Լ��䷺�ػ�(ͼ5G��S5C)�������ǰ������Ƶ�������IJ���Ҳ����NDRG3-VHL����õ���ǿ(ͼS5D)����ȱ���ͳ��������£��ڲ��ᴦ����ϸ����������Դ������������Ե�����NDRG3-VHL�����(ͼS5E)����ˣ����ǵó����ۣ�NDRG3-PHD2����ò���ϸ����������ˮƽ��Ӱ�죬��NDRG3-VHL�����������ˮƽ���������ƣ������ܵ���ˮƽ��Ӱ�졣
����������ȱ��ʱ�����Ĺ�������ֱ����NDRG3��ϣ�ͨ�����NDRG3-
vhl����ã������䷺�ػ��͵���ø�彵�⡣Ȼ����NDRG3���ػ��ڸ����������µ�ʧ���Ƿ�����������phd2�鵼��NDRG3���ǻ���������VHL���������ģ��д���һ���о���
ȱ��ʱNDRG3����raferk�ź�
Ϊ���˽�NDRG3��ȱ�������µķ��ӻ��ƣ�����ͨ��ʹ���ȶ�����NDRG3��GFP
shRNA��PLC/PRF/5ϸ�����������з�����Ѱ�ҿ��ܵ�NDRG3���ؼ�ø(ͼS6A)��NDRG3�Ľ�ѡ���Ե�����ȱ���յ���ERK1/2���ữ(ͼ6A��S6A)��Ȼ�����Ǽ����ERK1/2���εļ�ø�Ƿ���Ա�NDRG3���أ�����ȱ���յ���c-Raf
(at Ser338)��B-RAF1 (at
Ser445)�����ữ��SK-Hep-1ϸ���е�NDRG3�Ľ�������(ͼ6B)����Щ�����ʾNDRG3�����ڼ���raferk�ź�ͨ·�з�����Ҫ���á���ˣ������о��˲���NDRG3�����c-Raf���ữ��Ӱ�죬������λ�����c-Raf�ڳ����������������ữ��ͬʱ����ERK1/2���ữ(ͼ6C)��Ȼ����siRNAȱʧ��NDRG3�ļ�������ȡ�������ַ�Ӧ����һ���棬NDRG3(N66D)����λ����߶��յ���c-Raf��ERK1/2�����ữ(ͼ6C)�����⣬ȱ���յ���Դ��c-Raf��ERK1/2�����ữ��NDRG3��3��-UTR-targeting
shRNA���ƣ�����NDRG3(N66D)��������������(ͼ6D)������˫������ʵ�������NDRG3������c-Raf����ֱ�ӵ���������(ͼS6B)��
��ȱ�������£���λ�����c-Raf���߳�����Դ��NDRG3������������(ͼS6C)�����⣬�����⼤øʵ���У�HEK293Tϸ�������ĺ�NDRG3(N66D)�ĸ������߳����鵼������c-Raf�����ữ(ͼ6E)����Щ�������NDRG3ֱ�Ӳ�����c-Raf�����ữ��
ͼ����ͼfigs6
ͼs6��NDRG3��صĵ���ø�źŷ�������ͼ6���
��ʾ�����ı���
�鿴��ͼ�鿴�����ظ���ͼƬ����(PPT)
ͼ����ͼgr6
ͼ6NDRG3��ȱ���յ���raferk1 /2��������Ҫ��
��ʾ�����ı���
�鿴��ͼ�鿴�����ظ���ͼƬ����(PPT)
Ȼ�������о���ndrg3�鵼��c-Raf-ERK1/2���ữ������ѧ���塣ͨ������Դ�Ե������߳������������ǹ۲쵽����ȱ�������У�c-Raf-
ndrg3��������������ӣ�ͬʱc-Raf��ERK1/2�����ữˮƽҲ��֮����(ͼ6F)����һ�����ʾndrg3�鵼��c-Raf-ERK1/2���ữ������ȱ����Ӧ�����з������á�ͨ������LDHA��������IJ�������Ч������ȱ���յ���c-Raf��ERK1/2�����ữ��NDRG3���ı���(ͼ6G)��ͨ����Դ�ṩ�������Σ���������sildhai�鵼��c-Raf��ERK1/2���ữ�����������Ʊ���ĬMCT1�ı�������ϡ����⣬�����ǰ�����ǽͽ���ƻ�������Ч����c-Raf��ERK1/2�ĵ������ữ�Լ�NDRG3�ı��NDRG3���������һ����(N66D)(ͼ6H)�����֮�£�ȱ���Ȱ�������Ӱ����������Щ���������ȱ���յ���c-Raf��ERK1/2�����ữ����������IJ�������Ҫ�����ǽͽ⣬��NDRG3�������յ���raferkͨ·�������Ҫ�н顣
�����յ���ȱ��ϸ��������Ѫ������������ndrg3�鵼��ERK1/2����
��������о���ndrg3�鵼��rafe -
erkͨ·�����������������ȱ����Ӧ������ѧ����ԡ���Դ�ṩ�������������ֲ����ȱ��ʱLDHA��Ĭ�����Huh-1ϸ������ȱ��(ͼ7A)��Ȼ����ͨ��NDRG3�ĺĽ�ERK�źŵ�ҩ����ϣ�����鵼�����ȱ�ȡ����ͬ������Դ������ɻָ������յ���LDHA-knockdown
Huh-1ϸ���ڳɹ�ʵ���е�Ѫ��������������NDRG3�Ľ�ERK�����ֿ�������������(ͼ7B)�����ͬʱ��Ѫ�����ɱ�ǻ���ĵ��������LDHA�ó�������Դ������ָ�������NDRG3�Ľ�ERK���ƺ��ٴ��ж�(ͼS7A)��Ȼ�����Ǽ����ndrg3�鵼��raferkͨ·������������������������ԡ�Western
blot���������������������У����LDHA��/��NDRG3�������Ƶ�Huh-1ϸ���γɵ�����(ͼ4G��S4L)��������ģ�������(ͼ7C��S7B)��ȣ�����NDRG3(N66D)��������c-Raf��ERK�����ữ�����ϵ�������һ�¹۲쵽NDRG3(N66D)����������Ѫ�����ɱ�ǻ������Ҫ�����Щ����Լ�ͼ4��ͼ6�еĽ����������ȱ�������£��������ڴٽ�ϸ��������Ѫ�����ɷ���������Ҫ���ã���ȡ����ndrg3�鵼��c-Raf-ERK1/2ͨ·�ļ��
��
Ȼ������ͨ�����˸ΰ�(HCC)��������֯��ѧ�����������NDRG3�����ERK1/2���Ե��ٴ�����ԡ�NDRG3�����������м������������ϸ���ʺ���Ĥ�е�HCC��֯�пɼ��и߱���(ͼ7D)����103��ʹ��NDRG3��phospho-ERK1/2�������HCC�����У�25��(24.3%)NDRG3���ױ������ԣ���ERK1/2�����������(ͼ7D)����֮����Щ�������NDRG3���쳣���������������ķ�չ�Լ�ERKͨ·�IJ�������������ء�
����
����һֱ����Ϊ���ǽͽ�Ȱ������ֽ���ն˲��ֱ���������Ϊ�����Դ������Ѫ�����ɵ��յ������(Doherty��Cleveland,
2013)��LDHA������µ�������Ե���������������������ϸ��������(Fantin et al.�� 2006, Le et al.��
2010)��Ȼ���������յ������ﷴӦ�Ĺؼ����غͻ�����Ȼδ֪���������о��У�����֤����ndrg3�鵼�������źŵĴ��ڼ�����ȱ����Ӧ�е����á���ȱ��������������ˮƽ����ʱ��NDRG3���ı��ﱻ�߶��յ����Ӷ�������rafe
- erkͨ·���ٽ�Ѫ�����ɺ�ȱ��ϸ������������ˣ�NDRG3��Ϊһ�����ᴫ��������ȱ�������ķ�ʽ�������μ�ø�źţ�NDRG3- rafe -
erk��Ϊ�����յ���ȱ����Ӧ�ṩ���Ŵ�������
���Ƿ���NDRG3�ı�����HIFs�أ��ڵ���ˮƽ���������ξ��������������ȱ���ĺ��,�ٽ��ǽͽ��upregulation��LDHA������ȱ��������HIF-1�������Źؼ��Ľ�ɫ��Ϊ��л��Ӧ��һ����(Cassavaugh���ʲ���,2011)�����,�����źź��������ﷴӦ�ƺ��������HIF-1��-induced��л�ر��,����NDRG3�ؼ����ӡ����ⷽ��,���鲿��ȱ����Ӧ,�ر��Ƿ�����ȱ���ĺ��,����Ϊֹ�ѹ�����HIF-1������,��ʵ��,NDRG3-mediated����ֱ�ӿ����µ��źš��������Ľ����������NDRG3��ȱ���ڼ�HIF-1��֧�����ֿ�����(ͼ3��S3)�����,���ǵ��о�����,HIF-1����NDRG3�����γ�һ���������Եļ����ȱ����Ӧ,���·�Ϊ����ʱ���(ͼ7
e);�����ڽ�,����HIF-1��O2�������ź�,Ȼ��������ڻ���������������Ӧ�Է�Ӧ������л�ر��,�ں��ڽ�,�������������ź�NDRG3�Ļ���,�����Raf-ERKͨ·�յ���Ӧ����Ӧ�Գ���ȱ����
ȱ���źŵ�����- ndrg3 - rafe -
erk����ʾ��ȱ������IJ�������������������һ����ɲ��֣��ڳ�ʱ��ȱ�������´ٽ�Ѫ�����ɺ�ϸ���������Զ���,���ֹ���֮������HIF-1��-induced�����л�ر�̺�NDRG3-mediated�ź�ȷ��ϸ�����ٳ���ȱ��ȱ��������ʵ�������ܵ������������ͨ��,������������ϳɵĻ�ľ������ͨ��HIF-1��-mediated
upregulation�ǽͽ�,���,ͨ���ṩ����ϸ��������Ѫ������ͨ��NDRG3-mediated
c-Raf-ERK�źš���ˣ�ndrg3�鵼�������źſ���Ϊ�ֲ���֯�е�ϸ����ȱ���лָ��ṩһ���Ը�����Ļ��ƣ�������Ҫ�����ϸ�����źţ������ڷ����ڼ䡣���⣬NDRG3�鵼���ź�ת��Ϊϸ���ӱܳ�ʱ��ȱ���ṩ�˶�������ﰲȫ�㣬��ΪNDRG3����һ�����������ȶ���������ʹ��ϸ��������ʱҲ�����൱�ȶ���
��
Խ��Խ���֤�ݱ�������������ڰ�֢��չ�з��ӻ������ã���Ϊ����Ϊһ��������л��鵼�˰�֢ϸ���Դ�л�Ĺ������ã��������������жԼ���ϸ�����͵ķǰ�֢ϸ����������(Doherty��Cleveland,
2013)�����ǵĽ���������ǽͽ��������������Ҫ��Դ������ȱ���յ�NDRG3���ı����raferk�ļ����ϸ�����ǽͽ�������̶Ⱦ������ӣ���ˣ�����-
ndrg3 - raf -
erk�ἰ����Ѫ�����ɺ�ȱ��ϸ�������е����õķ��֣�����Ϊ�ǽͽ���Ͷ�֢�����������ṩ����Ҫ�Ľ��͡����ⷽ�棬������ܱ���Ϊ��һ�ֵ�һ��л�����Ϊһ�����ȼ�ϣ�һ�ֵ��������������Ƽ���һ���źŷ��ӡ�
����ȱ����Ӧ������Ҳ������ϸ������(Cassavaugh��Lounsbury, 2011)��ȱ���Ĵ����방֢����Ԥ������Ч�����й�(Jubb et al.��
2010, Semenza,
2004)�����ȱ���ѳ�Ϊ��֢���Ƶ���Ҫ�е㡣���ܵ����յ��������ⷽ�����ҪĿ��,����˵��ĵļ�����HIF���ܲ����Է�ֹ�����յ������Ľ�չ,��Ϊ�����о�����,����,HIF-independentͨ·�����յ����Ƶ�����ʱ(Mizukami
et al ., 2005��,Mizukami et al ., 2007��,Carmeliet et al ., 1998��,Rapisarda et al
., 2009;�μ������Ի��֧��ʾ��)����Щ�۲�����������ɹ��Ŀ�ȱ�����Կ�����Ҫ����hif����ͨ·��hif����ͨ·��ҩ�����(Mizukami et
al.�� 2007, Fong,
2008)������HIF-1���Ĺ�����ϵĿ�����,NDRG3�ƺ���ȱ���ķ�Ӧ��Ȼ��ͬ�Ĺ��ܵ��ڵĻ�������ת¼�����ݷ���NDRG3
HIF1A-depletedϸ����ȱ������ˣ���Щ�۲������Լ����о�����ʾ��NDRG3��ȱ����Ӧ�е����ã�����HIF��NDRG3����ϰ�������ڰ�֢�����зdz���Ч����NDRG3��HIFs����ʹ��ʱ���������������ƣ���֧���˸ò��ԵĿ�����(ͼ3H)��
����������NDRG3Ϊȱ���źŵ�ȱ�����������ṩ����Ҫ���Ŵ�֤�ݡ�NDRG3��ȱ��״̬�µĵ��غ��ܱ�����PHD2/VHLϵͳ�������������ķ�ʽ����hif�����ͷ�hif������ȱ����Ӧ�����,lactate-NDRG3-Raf-ERK�ź�ͨ·�����ṩ�ӳ���е�������������ϰ������ͻ��VHL(��Ѫ��ϸ����,��ϸ�������ȸ�ϸ������)(Maher
et al ., 2011)��PHD2(����erythrocytosis-3)(����et al .,
2006)�Լ�hypoxia-related�����Ͳ���������Ӧ(Cassavaugh���ʲ���,2011)��
A Lactate-Induced Response to Hypoxia: Cell
https://www.cell.com/cell/fulltext/S0092-8674(15)00264-0#articleInformation
��
NDRG3 lowers the metastatic potential in prostate cancer as a feedback
controller of hypoxia-inducible factors | Experimental & Molecular Medicine
https://www.nature.com/articles/s12276-018-0089-y
��
������:��л�ڰ�֢����ؼ�����
Lactate: A Metabolic Key Player in Cancer | Cancer Research
��
Franziska Hirschhaeuser, Ulrike G.A. Sattler, and Wolfgang Mueller-Klieser
Authors' Affiliation: Institute of Physiology and Pathophysiology, University
Medical Center of the Johannes Gutenberg University Mainz, Mainz, Germany
���������ǵ����պͻ������ᣬ��ʹ��������������(�������ǽͽ��WarburgЧӦ)�ǰ�ϸ����һ����ͬ��������һ��������ر��������β�������ȱ�����������������ο���Ԥ��ת�ƺͻ��ߵ��������棬�����ɼ��ͬʵ����о���ʾ�ġ�������ת������������صij���άϸ�����������յ������������ٽ��ģ���ΪǨ�ƴ����������Ļ��������᱾���Ѿ������ֿ����յ�ϸ����ϸ���ص�Ǩ�ơ����⣬�ͷ�����������Ũ�ȳ�����أ���ʾ������п�����������������л��������ϸ������õĽ����������������������й��ס����⣬���������˿����ϡ�������֢�Ͱ�֢�ķ�չ֮�����������������ϸ��ͨ�����������յ���VEGF��Ϊ��ֳ�ṩ�����������Ӫ�����Ӷ��γ��µ�Ѫ�ܡ���֮����ʵ�����������εĻ����Ƕ���������չ�Ĺؼ��������¼��������εIJⶨӦ�ý����һ�����ٴ����飬��ȷ�����ڰ�֢����ѧ�е�����ԡ���֢Res;71
(22);6921 - 5��AACR©2011��
��
����
Warburg��20����20����Ŀ������о������������о��ɹ�(1)֮���������ǽͽ�������л��������˴������о�������������Щ�о���Ҫ�����ڷ��������ϸ��������ϸ��ϵ��̼ˮ�������л���������ء����ͬʱ�������ķ�������Ҳ�ڲ��ϵ��ƽ���Ч��ҲԽ��Խ�ߣ����ƺ�����һ����Ҫ������:�������ޣ������������ƽ�⣬������������ȱ������ˣ���ȱ�����о����������֢���ߵ�ԭ��ͺ���Լ������Ʋ����������ԣ������������˶�������л���������о���������ȱ����ȣ������ǽͽ����������ѧ�����������ܵ���ѧ��ע��Ȼ�����й��ǽͽ���ػ�����ȫ��70%�����֢�й�����ķ���(2)���Լ�ʹ��18f
-�����������ǵ������ӷ���ϲ���Ӱ��(positron emission
tomography)���ð�ϸ���������ǵ���ȡ����������������ϵķ��֣�ʹ��������ٴθ��ˡ��籾������������ܵ����ٴ������а�֢��Ϻ�������õĸ��ơ�������Ϊ��������������л����ؼ����õĹ۵���ͼ1��ʾ��
ͼ1
˵���������ǰ�֢�Ĺؼ����ء�DC,��ͻϸ��,EC,��Ƥϸ��;GLUT,������ת����;IL,����;HAT,�鵰��������ת��ø;LDH,��������ø;MCT,������ת����;PEP,����ϩ����ͪ��;PGAM,�������������λø;PPP����������;��;��������������;TAF,������س���άϸ����
���������еĴ�лת��
����ϸ���Ķ���ת�����´����ʵ����������ȱ����������������ȡ�������γ����ӡ��ձ���ܵ���,����������ϸ������ȱ�ݵĽ��,�°��ĸı�,���ȵ��ǽͽ�ø�ʹ�л����ת�˵���(3)�����������������ƻ�������л���������ữ(OXPHOS)������ϸ���ǽͽ�ĸı�,��myc��NF-��B��һ�ֵ���ø/����øB,��Ƥ��������(EGF)���ȵ�������������I,phosphoinositol
3��ø(PI3K)��mTOR
Kirsten�����������°�������ͬ����(��˹��),�����ø(AMPK)�͵����յ�����1��(HIF-1��;�ο����ס�3��4).������°������ѱ�֤���ɴ̼�����鵼�ǽͽ�Ȱ������ֽ�ĵ��Ļ���ת¼������ϸ����Ӧ�Ĺؼ��ɷ�HIF-1��,ͨ��ͨ��prolyl�ǻ�ø�ͷ�Hippel-Lindau�������ϸ������Ũ�ȿ��ơ�ע��,HIF-1��ͨ���ǽͽ�IJ���,����ͱ�ͪ�ᣬ�����ڳ����������ȶ�,���������ת¼����HIF-1���Ļ���(5)�����,���ϻ���(1)�۲�,HIF-1���ڻ���ı���ᵼ�������ж�����������һ����ǿ���ǽͽ�ͨ�������ӡ���ˣ�ʵ������������۵�����������ȱ�������(6)��Yaromina��ͬ��(7)��������������ˮƽ�ϣ�������Ũ����ȱ���ء�(ȱ���������Ǵ�л�������������������Ҳ�������������DZ��ĵ���Ҫ���⡣)HIF-1�����ðе����Ĥת�˵��ף���������ת�˵���1
(GLUT-1)��һԪ����ת�˵���4 (MCT-4)�������ֵ����ܱ�֤�㹻�������ǽ���ϸ�������ܱ�֤���۵�������ڳ�ϸ�������⣬������������øA
(LDH-A)������ǿ��NAD+���ɣ����������ǽͽ��ATP�IJ�������ͪ������ø��ø1��HIF
-1�����Լ����Լ��ɴ˵��µı�ͪ������ø�������ʧ������������ữͨ���Ľ��͡����ܸ�ת���ʵı�ͪ��ת��Ϊ������ǽͽ�;��,һЩ��ͪ�ỹ�д�������(TCA)������������������ѧ������ϳɵ�Ŀ��(4)��������ѭ������������;��(PPP)���Ա�������ϸ����ֳ�ĸ�ǰ������,ͬʱ������Χ������֯�������л��塣���⣬PPP����NADPH��Ϊ��������Ӧ���н飬����ϸ��������������(3)�������ǽͽ⣬�Ȱ������ֽ�����һ����Ҫ������������Դ���ƺ��������������ϸ����������ۡ��Ȱ������ֽ�ܴٽ���ֳϸ���д���ӵĺϳ�(4)����һ��������Դ�����������Եı�ͪ�ἤø(PK)
M2�����ܽ�����ϩ����ͪ��(PEP)ת��Ϊ��ͪ�ᡣȻ��,PEPҲ����Ϊһ�������߶��������������λø1
(PGAM1),�γɵı�ͪ��(8)������PKM2�Ļ���ܵ���˵,���ƺ��������ǽͽ�;���ؼ�øת����Щø����̥��ѹ��֧������Ĵ��ģ��ֳ(9)��һ����ͬ������
��������ݱ������ǽͽ�Ľ�������방ϸ��������صı����Ŵ������йء��ر���Buchakjian��Kornbluth(10)�����˱�ͪ����鵰������ø(HAT)���鵰��ȥ����ø(HDAC)��Ӱ�죬�����ǽͽ�ø��ת�˵���ת¼���ӡ���ˣ��ǽͽ�Ĵ�л����ܲ����˱����Ŵ�������·������һ��·Ŀǰ��֪֮���٣���Ҫ����һ������н�һ�����о���
��������ݱ������ǽͽ�Ĵ�лת��������������������ںͻ����¼���Schafer����ͬ��(11)���о����������Her2/neu+�������ٰ�ϸ���ǽͽ���ϵ���ͨ����ֹEGF������µ��Ӷ�ά��PI3Kͨ·�ļ��Ϊ���ṩ�������ơ�Ong������ͬ��(12)���֣���Ƕ���ճĤ��ȣ���ǰ��Ϣ���Լ��ٰ����ߵ�����������������ӡ�һЩ�������ڷ��ӵı任,��NF-��B��������֢��Э��������������Ϊ��֢�ǡ����Ϲ��ȵ��˿ڡ��Ĺ۵��йء����⣬�������֯�˿����Ϲ��̵Ļ���Ҳ�ǰ�֢�����ͽ�չ����Ҫ�������ػ�������(13)�����᱾����Ϊһ�����ڵ���֢���ʣ�����Tϸ���;���ϸ����������İ�ϸ������(IL)-17A���Ӷ��ٽ����������е�������֢(14)��
��
������������ݵĹ���
����������һ����Ҫԭ��������ϵͳ���������쳣ϸ����������о��Ѿ���ϸ����������ϸ���ļ������ݻ��ƣ��������Ʒ��ӵ��ϵ�����������ϸ�����ӵIJ������̼����ӵ��µ�(15)��������Щ�����⣬������лҲ�ںܴ�̶��ϲ������������ݡ����������о����ְ�����������Ƶ���ϸ������ͻ״ϸ��(DC)�ķֻ�����������ͻ״ϸ��(16)��ϸ������Tϸ��(17)�ͷŵ�ϸ�����ӣ��������ǿ�������Ӧ�Ĺؼ������߷���������ʾ���������������ܰ�ϸ��,�������ȱ�����߷�Ӧ������Ҫ�ǵ��͵�MHC-1��������ؿ�ԭ���ڵ�ʶ���ʧЧ,���෴,���ǹ�����Ӧ������ϵͳ�����ˡ�
��
�����Tϸ���������ǽͽ�Ϊ��Ҫ������Դ(17)��������ϸ����ϸ����ռ��ͷŴ�������ʱ������ϸ����������������ᣬ��Ϊϸ������ķ���������ϸ������Ũ�ȵı�ֵ�����գ���ϸ�����ܱ���������Ϣ������ϸ������ͨ��MCTs�ķ��ڰ�����H+�����䣬ϸ����pHֵ�Ľ��͵���ϸ������Tϸ�����ܵĽ���(17,18)���෴������Tϸ��(Treg)�ƺ��������������������Ӱ�죬��Ϊ�����в�ͬ��������л������֬��������(19)������Խ���Ϊʲôʵ��������������ϸ�������ʼȲ���Ԥ�⼲���Ľ����Ҳ����Ԥ��ߵ����档
�����ϸ��Ǩ�ƵIJ�ͬӰ��
�ھ����Boyden
chamberʵ���У���Դ������ļ��뵼���˶��ְ�ϸ������Ũ�ȵ����Ǩ������(20)����������������ʵ��������ص�����ˮƽ��,0-40����/
L)�����⣬����鵼������ϸ���˶���ǿ���������ڵ�ϸ���˶��У����һ�������ͨ����ʱ��������ǿ�ƵĴ���Ǩ����(20)�����������յ����źŵ���ˮƽ�仯���伤��״̬,���1-integrins�Ѿ��м��ء����ӻ��Ʋ��������ϸ�����Ե�Ӱ�컹����������������֧��TGF-��2�ź�ͨ·��һ������ЧӦ��ذ�ϸ��Ǩ�Ƶĵ�����(21)��
Goetze��ͬ��(20)�����방ϸ����ͬ��ʵ�����ã�������Դ������ʼ�����Ƶ���ϸ����Ǩ�ơ���һ���ְ���һ��Ũ������ϸ��������il -
6��TNF-���ͷŵļ���,����Ҫָ�����ǣ�������ʵ������У�pHֵ����������7.2���¡�
Boyden
chamberʵ�����������ͨ����Ƥϸ��(EC)�̼�VEGF�����ɣ�������ǿ��Ǩ�ƣ���������O2�����²��������յ���Ѫ������(22)�����,Vegran������ͬ����(23)�������־�Ϸ�����һƪ����,���DZ�ʾ,����������ECs
MCT-1����NF-��B���IL-8�ı������С��������ֲģ�ͣ����Ƿ�������ϸ��ͨ��MCT-4�ͷ��������Դ̼�il -8������Ѫ�����ɺ�����������
���ӵ������ij���άϸ���е������������������IJ�����������CD44�ı������ߣ�CD44��һ�ֿ�Ĥ�ǵ��ף���ϸ��������Ҫ�����������塣����Χ�Ļ���������������س���άϸ��(TAF)�����������ᣬ�ṩ��һ���ٽ���ϸ���������˶��Ļ���(24,25)��
���������ԭ���������е��ٴ�����
��2000�꣬����ʵ���ҵ����ݷ����ڱ����ϣ���ʾ�ڳ���9�������ڼ䣬ԭ���Թ�����������������뻼�������ʳʸ����(26)����������֪�����ǵ�һ��������֤�������ٴ������£����������л�방֢����Ϯ��������ء�������Ȼ������LDHø��Ϊ���ᴫ���������������֯�д�л��Ũ�ȵ����ݡ�����ø����������ӫ����ø�йأ����ͷŵ�����ӫ��ǿ�����л��Ũ�ȳ����ȡ�ʹ���ʵ��ı�,һ���˿���У���ź��ڵ�λ��֯Ũ��,��Ħ��ÿ����֯(��mol
/
g)���ڱ�����֯���յ�����ⷴӦ��ʹ�ø�����֯����֯ѧ�ṹ��¼���źų�Ϊ���ܡ������ϣ��յ���л��������(imBI)���������ͽṹ��صļ����֯�еĴ�л�����������ˮƽ�ϵķֱ��ʡ�������Щ���ص����ԣ�imBI�ڶ���������е�Ӧ��Ϊ������ת����֢�о��ṩ���µ���Ϣ������������
�ڷ������������ڳ���������������ز��˵���������ݺ�,һ��ͷ������״�����ߵĶ����о�������������ǰ���Ậ������ܻ��ƺͷ������ƺ��������������ʳʸ����(25��27)�����⣬�����о�������������Ậ����Զ��ת�Ƶķ����ʳ����������(25)������Щ�о��з��֣���ͬ���ںͷּ��������ڵ�������Ũ�Ȳ���ܴ��ڷ�����3��ʵ���У���������л��(��ATP��������)��ȣ�����������������Ũ����ʾ���dz���������ں�������ı仯��Ȼ�������������εı仯ԶԶС��������졣����imBI�������������������۵��ٴ�����ԡ�Ŀǰ����û��һ�ַ������Լ�������ͬʱ�ṩ���������ơ����ң�Ŀǰ���������Щ����������Ρ�����ͨ�����������Ժͷ������Է�����⡣Ȼ���������һЩ���������о�����������Ķ�������DZ�ڵ��ٴ����塣�����������Ƶ��о��У�Saraswathy������ͬ��(28)���֣����ӴŹ����������ĸ�������ǿ��������Խ���ĸϸ�������ߵĵʹ�����йء�Park������ͬ��(29)������ֲ���˽���ĸϸ������13C����ˮƽ���Ը��������Ĵ�����ԡ������о�ʹ����13C����ѧ��߷ֱ��ʵĻýǷ�˿����(30)��
�ڰ����л����ǰ��������Ӧ�����ᶨ���������������ṩ�йذе������ԡ�������Ӧ���ٴ�������м�ֵ����Ϣ���������Ŀǰ�ٴ��о���ʵ�������������������Ҫ�����á�
����Է���ֿ��Ĺ���
����һ���о���(�������ǵ�ʵ����)��ʵ�������Ľ��������1000�������HNSCC��������ֲ������������Ũ�������ֿ������(31)����������Կ��������ڣ����ٲ��֣�����ǰ���о��н�ʾ������Ŀ���������(32)�����������ƣ����������һЩ����ҩ��յ�����Ӧ����Ŀ��ϸ����������(ROS)�Ĺ��������ᵼ��DNA��RNA���ˡ�֬�ʹ������ͻ����鲻�ȶ��ԡ��������ǹ̶������յ���DNA�����������;��ˣ���������(��������)�Ļ��ۿ����յ�����ǿ�Է���ĵֿ��������ܵ��»�ѧ�ֿ�(33)�����ڶ����ڽ��ܻ��ƻ���ƺ�����ˮƽ�½�(34)����˼�����������������л�������������Ԥ�����Ʒ�Ӧ��
��֮��ͨ���ǽͽ����ı俹������л�����ˮƽ���Ӷ��ı�ROS�����������ܻᵼ�¸��õ����Ʒ�Ӧ��
������δ��չ��
����ϸ�����д�лת���������ٽ�ϸ�������ͷ��ѵ��м��塣���ƺ���һ���dz����ڵ�(������������)�°��¼���������Ŀǰ�۲쵽�Ĵ�����������ˡ�������Ŀǰ��֪ʶΪ���������ھ�ȥ�����ǽͽ������������е����õó�ȷ�еĽ��ۻ�Ϊʱ���硣Ȼ����Խ��Խ�������֧�ֽ���һ������Ϊδ��������л�����о����ݼ����衣��ͼ������֢�¼�����л�쳣������ı�����Զ��������������еġ����͵����⡱��ʵ����֢�о���һ�������˷ܵ���ս��
��ȻһЩ�Ŵ��������Ͳ������������ѱ�ȷ��Ϊ���¸������������߶ȶ��Ե�ԭ��Ϊʲô������ͬ����������������֯�����Ậ���ϱ��ֳ����˲�������һ���ա��������Ǹ�����δ���о�����һ��ս��
����ȱ����Щ֪ʶ������������л��ת���о��Ѿ�ת�Ƶ�����֢�о���һЩ��������ת�Ƶ��ٴ������С�Tennant������ͬ��(35)��д�˴������ڲ���������л���ٴ�����ѧ���飬ֱ��III�ڡ�����������������о���ԱΪ����������л��Ϻ����ư�֢�Ĺ�ͬŬ���������ס�
Lactate: A Metabolic Key Player in Cancer | Cancer Research
https://cancerres.aacrjournals.org/content/71/22/6921.long
��
���������һ�����͵���������ø���Ƽ���ͨ��Ӱ�첻ͬ���ź�ͨ·���յ���ͬ�ǽͽ�̬�ȵ������ٰ�ϸ������
Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human
breast cancer cells with different glycolytic attitude by affecting distinct
signaling pathways
ժҪ
������ֵ�һ����������ø���Ƽ���ͨ����ֹ�ǽͽ��ATP�IJ�������ֹ��ϸ������ֳ����ʵ���Ŀ�����о��û���������ٰ�ϸ��ϵ��Ӱ�죬��Щϸ��ϵ�ɲ����������IJ�ͬ��������:MCF-7(�߷ֻ���)��MDA-MB-231(��Ϯ������������)��MCF-Tam(���л���������������Ե�MCF-7����)��
���ǹ۲쵽��Щϸ��ϵ��������л���������졣��MCF-7ϸ����ȣ�MDA-MB-231��MCF-Tamϸ�������ֳ��ϸߵ�LDHˮƽ����������ȡˮƽ�������ֳ��ϵ͵ĺ����������ܴ�����Щ���죬��GFҲ�����Ƶ������������á���һ�������ͨ����MDA-MB-231��MCF-Tamϸ���з���һ�ֽṹ�Լ����Ӧ����Ӧ�����ͣ���Щϸ��������Ԥ������������̬����һ��֤������ͬ���ź�ͨ·������GF�Ŀ���ֳ���á�MCF-7ϸ�������ǹ۲쵽ER��-mediated�ź�����ĵ���ϸ�����档�෴��MCF-Tam��MDA-MB-231ϸ�������������ƺ���������Ӧ��������ɵġ�ϸ���������ձ�������յ�������
���ھ����������Ժ�/��ѧ���Ա��͵����ٰ���ʽ���ٴ�����ԣ����ǵĽ����ʾGF�����Ի���������ϸ��Ҳ�����Ƶ����ã��������һ�����о�����֤���ֻ������ڸ������ٰ������е�DZ����
Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of
human breast cancer cells with different glycolytic attitude by affecting
distinct signaling pathways
Abstract
Galloflavin (GF), a recently identified lactate dehydrogenase inhibitor, hinders
the proliferation of cancer cells by blocking glycolysis and ATP production. The
aim of the present experiments was to study the effect of this compound on
breast cancer cell lines reproducing different pathological subtypes of this
tumor: MCF-7 (the well differentiated form), MDA-MB-231 (the aggressive triple
negative tumor) and MCF-Tam (a sub-line of MCF-7 with acquired tamoxifen
resistance).
We observed marked differences in the energetic metabolism of these cell lines.
Compared to MCF-7 cells, both MDA-MB-231 and MCF-Tam cells exhibited higher LDH
levels and glucose uptake and showed lower capacity of oxygen consumption. In
spite of these differences, GF exerted similar growth inhibitory effects. This
result was explained by the finding of a constitutively activated stress
response in MDA-MB-231 and MCF-Tam cells, which reproduce the poor prognosis
tumor forms. As a further proof, different signaling pathways were found to be
involved in the antiproliferative action of GF. In MCF-7 cells we observed a
down regulation of the ER��-mediated signaling needed for cell survival. On the
contrary, in MCF-Tam and MDA-MB-231 cells growth inhibition appeared to be
contributed by an oxidative stress condition. The prevalent mechanism of cell
death was found to be apoptosis induction.
Because of the clinical relevance of breast cancer forms having the triple
negative and/or chemoresistant phenotype, our results showing comparable effects
of GF even on aggressively growing cells encourage further studies to verify the
potential of this compound in improving the chemotherapy of breast cancer.
Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of
human breast cancer cells with different glycolytic attitude by affecting
distinct signaling pathways - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0928098712003235
��
ûʳ����Ŀ������ü�����ӻ��Ƶ��о�չ��
��֢��ȫ��ڶ�������ԭ�����Ƕ����Ϳ���ҩ�������һֱ�ܴ�ѧ����̽��������Ȼ�����컯�������˷���һ���⡣ûʳ����(GA)��һ�ַ��ᣬ����������Ŵ�ҽҩ�С������п��ס����������������Ϳ��������ԡ����������˳�ù�ؼ���������Ŀ������ԡ������˳�ù�ضԶ��ְ�ϸ�����������ʵ��������ǰ���о�����,GA�Ŀ���������ͨ����ͬ�Ļ���(���յ�ϸ��������ش��Ļ�����(ROS),���ڵ����Ϳ���������,���ƺʹٽ��°�����,���ƻ��ʽ�������ø(MMPs)��ϸ����������ȡ�������͵İ�֢�о�������������GA�������������Ϊ�������ư�֢����Чҩ�Ҳ������������ҩ������ʹ�ã������Ч�ʡ�Ȼ������Ȼ��Ҫ����Ķ���ģ�ͺ������ٴ��������ƹ㲢��GA����������������ҵ�г���
allic acid: prospects and molecular mechanisms of its anticancer activity Cancer is the second leading cause of death worldwide. There is always a huge demand for novel anticancer drugs and scientists explore various natural and artificial compounds to overcome this. Gallic acid (GA) is one of the phenolic acids found in many dietary substances and herbs used in ancient medicine. It possesses antiinflammatory, antioxidant, antiviral and antibacterial properties. The present review summarizes the anticancer activity of GA and its derivatives. Various in vitro and in vivo experiments of GA against a variety of cancer cell lines were reported. The previous studies show that the anticancer activity of GA is related to the induction of apoptosis through different mechanisms like generation of reactive oxygen species (ROS), regulation of apoptotic and anti-apoptotic proteins, suppression and promotion of oncogenes, inhibition of matrix metalloproteinases (MMPs) and cell cycle arrest depending upon the type of cancer investigated. Conclusively, GA and its derivatives may be considered as a potent drug for cancer treatment alone as well as in combination with other anticancer drugs to increase the efficiency of chemotherapy. However, there is still a need for more experimentation in knock-out animal models and human clinical trials to promote and place GA and its derivatives on the commercial market.
��
The Rate of Oxygen Utilization by Cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3147247/
��
��
Targeting tumor perfusion and oxygenation to improve the outcome of anticancer therapy1
Radiotherapy and chemotherapy are widespread clinical modalities for cancer treatment. Among other biological influences, hypoxia is a main factor limiting the efficacy of radiotherapy, primarily because oxygen is involved in the stabilization of the DNA damage caused by ionizing radiations. Radiobiological hypoxia is found in regions of rodent and human tumors with a tissue oxygenation level below 10 mmHg at which tumor cells become increasingly resistant to radiation damage. Since hypoxic tumor cells remain clonogenic, their resistance to the treatment strongly influences the therapeutic outcome of radiotherapy. There is therefore an urgent need to identify adjuvant treatment modalities aimed to increase tumor pO2 at the time of radiotherapy. Since tumor hypoxia fundamentally results from an imbalance between oxygen delivery by poorly efficient blood vessels and oxygen consumption by tumor cells with high metabolic activities, two promising approaches are those targeting vascular reactivity and tumor cell respiration.This review summarizes the current knowledge about the development and use of tumor-selective vasodilators, inhibitors of tumor cell respiration, and drugs and treatments combining both activities in the context of tumor sensitization to X-ray radiotherapy. Tumor-selective vasodilation may also be used to improve the delivery of circulating anticancer agents to tumors. Imaging tumor perfusion and oxygenation is of importance not only for the development and validation of such combination treatments, but also to determine which patients could benefit from the therapy. Numerous techniques have been developed in the preclinical setting. Hence, this review also briefly describes both magnetic resonance and non-magnetic resonance in vivo methods and compares them in terms of sensitivity, quantitative or semi-quantitative properties, temporal, and spatial resolutions, as well as translational aspects.
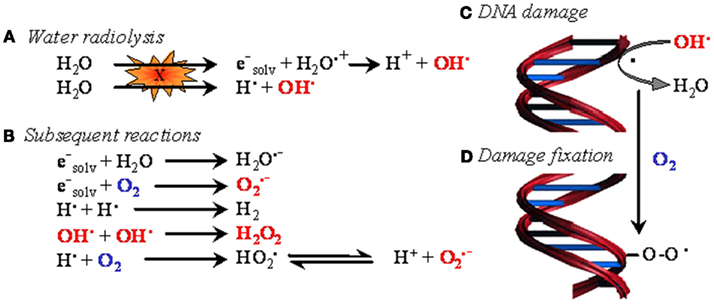
Figure 1. The oxygen enhancement effect in radiotherapy. In biological tissues, irradiation primarily induces water ionization and destabilization, leading to the formation of reactive radical species (A). These species then react with neighboring molecules to yield reactive oxygen species (ROS) (B), among which the hydroxyl radical is believed to be the most cytotoxic. When generated in the proximity of DNA, hydroxyl radicals and, to a lesser extent, other less energetic species attack DNA (C). The resulting formation of a DNA radical is readily reversible. However, in the presence of oxygen, DNA damage can be stabilized through oxidation of DNA radicals, eventually leading to the formation of DNA peroxides (D). In oncology, oxygen-dependent DNA damage fixation is known as the ��oxygen enhancing effect�� of radiotherapy.
��
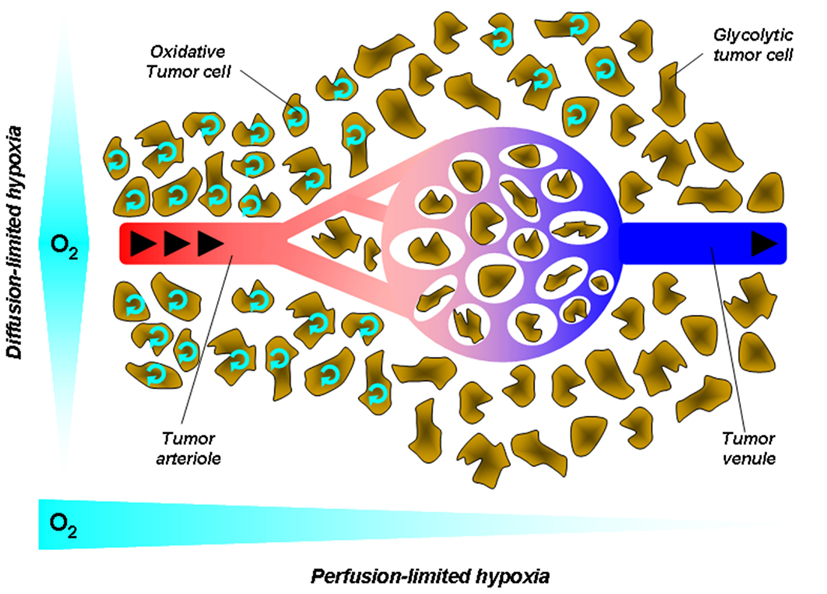
Figure 2. Tumor hypoxia. Hypoxia in tumors results from a mismatch between the oxygen supply by poorly efficient blood vessels and oxygen consumption by metabolically overactive tumor cells. This simplified cartoon depicts two main forms of hypoxia. Diffusion-limited hypoxia refers to a gradient of oxygen deprivation from the nearest perfused blood vessels toward tumor cells at increasing distances from this vessel. It originates from high-rate of oxygen extraction though layers of cells within a loosened vascular network. Perfusion-limited hypoxia refers to oxygen deprivation along the vascular tree from the tumor margin toward the tumor core. Poor oxygen delivery has many causes in tumors, including high-rate of oxygen extraction at the tumor margin, decreased red blood cell deformability, and stacking, increased blood viscosity due to water extraction, vascular disorganization, and angiogenesis. Arrows represent the blood flow.
��
Hypoxia, a partial pressure of oxygen (pO2) below physiological needs, is a limiting factor affecting the efficiency of radiotherapy. Indeed, the reaction of reactive oxygen species (ROS, produced by water radiolysis) with DNA is readily reversible unless oxygen stabilizes the DNA lesion. While normal tissue oxygenation is around 40 mmHg, both rodent and human tumors possess regions of tissue oxygenation below 10 mmHg, at which tumor cells become increasingly resistant to radiation damage (radiobiological hypoxia; Gray et al., 1953). Because of this so-called ��oxygen enhancement effect�� (Figure 1), the radiation dose required to achieve the same biologic effect is about three times higher in the absence of oxygen than in the presence of normal levels of oxygen (Gray et al., 1953; Horsman and van der Kogel, 2009). Hypoxic tumor cells, which are therefore more resistant to radiotherapy than well oxygenated ones, remain clonogenic, and contribute to the therapeutic outcome of fractionated radiotherapy (Rojas et al., 1992).
��
Tumor hypoxia occurs in two ways: chronic hypoxia (or diffusion-limited hypoxia), and acute hypoxia (or perfusion-limited or fluctuating hypoxia; Figure 2). Chronic hypoxia has classically been thought to result from long diffusion distances between tumor vessels as the consequence of the more rapid expansion of tumor cells than that of the supporting vasculature (Vaupel et al., 1989). It is now well established that steep longitudinal gradients of pO2 along the vascular tree, as opposed to radial diffusion of oxygen, can largely contribute to deficiencies in tumor oxygen supply (Dewhirst et al., 1999).
Hyperthermia: Combining Provascular and Oxygen Consumption Effects in a Single Treatment Hyperthermia is a potent adjuvant therapy with radiotherapy and chemotherapy, and the perfect illustration of a strategy combining transient, local vasodilatation with the inhibition of tumor cell respiration. The heat treatment consists of elevating the temperature of tumors to a supra-physiological range of 40�C45��C at which tumor reoxygenation occurs with limited skin toxicity. Hyperthermia induces a graded response in tissues characterized by decreased oxygen consumption at temperatures ��40��C, vasodilatation between 41 and 41.5��C, and vascular damage above 42��C. Although direct tumor cell killing was demonstrated in vitro at higher temperatures, long-term tumor control has never been demonstrated using hyperthermia as the sole treatment modality. Vasodilation only modestly contributes to tumor reoxygenation at the low thermal doses. Increased pO2 rather primarily results from changes in oxygen consumption in the target cells, and at least two different processes have been identified to contribute to this response. It is now well demonstrated that an important target of heat is proteins among which enzymes of the respiratory chain are more sensitive to heat inactivation/denaturation than glycolytic enzymes (Lepock et al., 1987; Kelleher et al., 1995). But the inhibition of mitochondrial respiration by heat lasts longer than the turnover time of respiratory enzymes, suggesting the existence of an additional mechanism. Dewhirst in collaboration with our team (Moon et al., 2010) recently demonstrated that mild hyperthermia activates the transcription factor hypoxia-inducible factor 1 (HIF-1) through an hypoxia-independent mechanism involving the sequential activation of Extracellular signal-Regulated Kinases (ERK) by heat shock, ERK-induced upregulation of the expression of the Nox1 subunit of NAD(P)H oxidase, increased ROS production by NAD(P)H oxidase, ROS-induced HIF-1�� protein stabilization, and, ultimately, HIF-1 activation. HIF-1 target genes include most glycolytic enzymes and transporters as well as major pro-angiogenic molecules such as VEGF. Among these genes, we showed that pyruvate dehydrogenase kinase 1 (PDK1) largely mediates the inhibition of mitochondrial respiration by heat in tumor cells through inhibiting pyruvate dehydrogenase (PDH), i.e., the enzyme coupling glycolysis to the tricarboxylic acid (TCA) cycle (Moon et al., 2010). Furthermore, consistent with the increase in VEGF expression that we also observed in heat-treated tumors, we documented an increased vascular density in perfused tumor areas where oxygen is extracted from the blood. Tumor reoxygenation by mild hyperthermia is thus a multifaceted process involving the combination of decreased O2consumption by tumor cells and increased O2 delivery by blood vessels. This and the fact that reoxygenation occurs at thermal doses lower than those inducing vascular damage justifies the use of mild hyperthermia as a combination treatment notably with radiotherapy. Although several clinical trials have confirmed that combining heat and radiotherapy is indeed associated with better patient treatment outcome (Brizel et al., 1996; Jones et al., 2003, 2005; Vujaskovic et al., 2003) the future clinical development of hyperthermia strongly relies on designing tools allowing for homogeneous thermal dose distribution and improving imaging techniques able to correlate thermal maps of tumors to the clinical outcome of patients (Dewhirst et al., 2010).
Frontiers | Targeting Tumor Perfusion and Oxygenation to Improve the Outcome
of Anticancer Therapy1 | Pharmacology
https://www.frontiersin.org/articles/10.3389/fphar.2012.00094/full
Front. Pharmacol., 21 May 2012 | https://doi.org/10.3389/fphar.2012.00094
��
J Cancer 2016; 7(7):817-822. doi:10.7150/jca.14274
Warburg Effect - a Consequence or the Cause of Carcinogenesis?
Slobodan Devic
Department of Radiation Oncology, SMBD Jewish General Hospital, McGill
University, Montr��al, Qu��bec, Canada.
abstract
��
��
Ever since its discovery (1924) the Warburg effect (aerobic glycolysis) remains an unresolved puzzle: why the aggressive cancer cells ��prefer�� to use the energetically highly inefficient method of burning the glucose at the cellular level? While in the course of the last 90 years several hypotheses have been suggested, to this date there is no clear explanation of this rather unusual effect. Even though it is commonly assumed that Warburg effect is a consequence of carcinogenesis, yet another hypothesis could be brought up that the cellular switch to aerobic glycolysis may represent the very point in time when a normal cell becomes cancerous. Furthermore, this switch may happen at the point where the fate of pyruvic acid is determined, caused by the inadequate supply of enzymes that promote citric as opposed to lactic acid cycle. Currently, few clinical observations, like low cancer incidence in Type 1 diabetes mellitus and increased cancer incidence in people on high carbohydrate diets might be called upon to support such hypothesis.
��
Economy of cellular energy balance
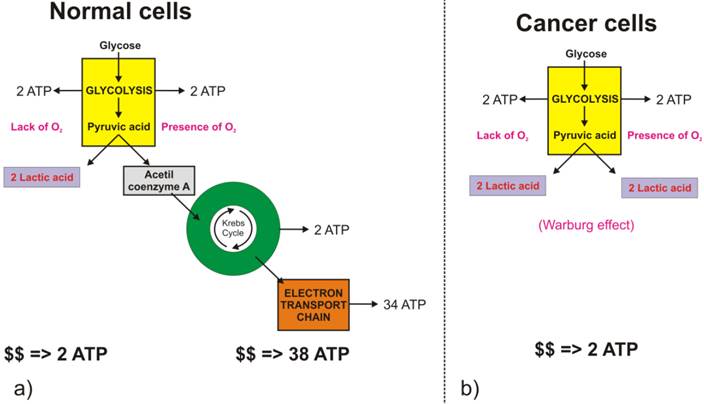
The full oxidation of one glucose molecule (oxidative phosphorylation) within a cell in the presence of oxygen produces 38 molecules of adenosine-three-phosphate (ATP), which in turn represents the essential cellular fuel (Figure 1.a). The first step (Glycolysis, occurring in cytoplasm) of glucose cellular respiration produces only 2 ATP molecules and ends up with production of two molecules of Pyruvic acid. If a given cell has access to oxygen, the Pyruvic acid will be converted to Acetyl-coenzyme A, which enters the Krebs cycle (citric acid cycle, occurring within mitochondria) followed by the electron transport chain process (occurring on the inner mitochondrial membrane) that creates most of the ATP molecules. It is the very last step within electron transport chain that needs oxygen to collect the terminal electron from the last cytochrome (cyt a3) and become a nascent O- to pick up 2 H+ and create one of the byproducts of aerobic cellular respiration - a water molecule. On the other hand, if the cells reside under hypoxic conditions, the pyruvic acid is not converted into acetyl-coenzyme A, but into a lactic acid - the process termed anaerobic cellular respiration (lactic acid cycle). In the latter case, the net energy balance is only two ATP molecules making the anaerobic glucose metabolism energetically highly inefficient process.
While the lack of oxygen at the cellular level can occur at times of excessive physical activity (resulting in subsequent muscle pain), it also represents a hallmark of highly invasive and fast-growing cancers [[1]]. As the cancer cells multiply, their fast multiplication rate outgrows the angiogenesis so much so that while the glucose access to fast-growing cells could be sufficient the lack of blood vessels disrupts the level of oxygen needed for the full glucose oxidation. Such a microscopic picture defines one of the fundamental properties of the cancer microenvironment that forces malignant cells to metabolize glucose through the lactic acid cycle. Looking at chemical equations between aerobic and anaerobic glucose metabolism (Figure 1) one may conclude that cancer cells (as well as healthy ones) under hypoxic condition would need 19 times higher uptake of glucose to maintain the same metabolic level as well-oxygenated cells. This would, in turn, mean that the appropriate quantitative analysis of FDG-based PET images could help in pinpointing hypoxic segments (by abundance only [[2]]) of the tumor by solely looking into very high uptake values, which would eventually be some 20 times greater than in well-oxygenated cancer cells. However, the isolation of the hypoxic target volumes is far from being that simple.
Warburg effect, or aerobic glycolysis - hallmark of invasive cancers Apart from the fact that acute hypoxia in tumors develop as soon as one moves few hundred microns from the blood vessels, yet another important fact prevents FDG being an ideal hypoxia marker - the Warburg effect. Recently, interest in tumor metabolism has been revived partly as a result of the widespread clinical application of PET using FDG. FDG-based PET imaging has confirmed that most primary and metastatic cancers show a significant increase in the glucose uptake when compared to normal tissues.
��
Warburg effect, or aerobic glycolysis - hallmark of invasive cancers Apart from the fact that acute hypoxia in tumors develop as soon as one moves few hundred microns from the blood vessels, yet another important fact prevents FDG being an ideal hypoxia marker - the Warburg effect. Recently, interest in tumor metabolism has been revived partly as a result of the widespread clinical application of PET using FDG. FDG-based PET imaging has confirmed that most primary and metastatic cancers show a significant increase in the glucose uptake when compared to normal tissues.
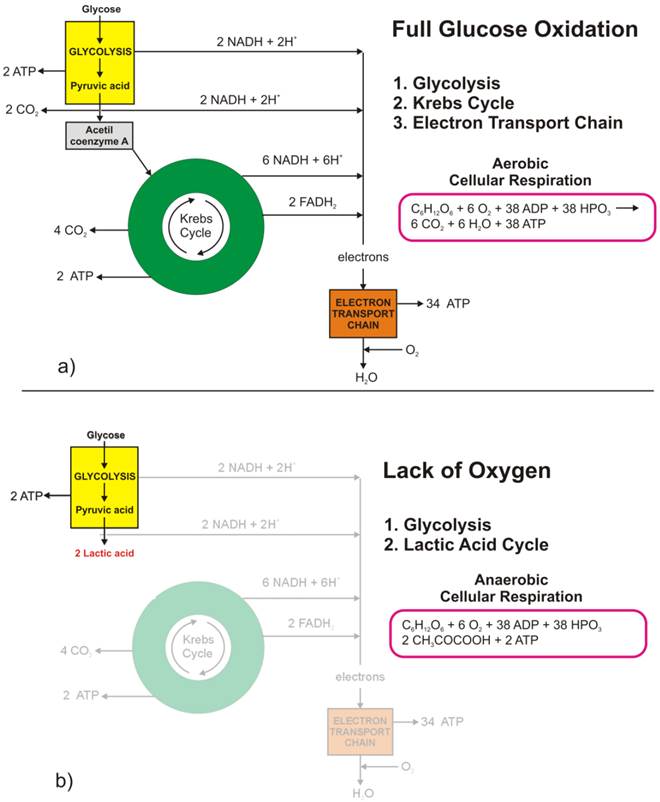
Figure 1 Glucose metabolism at the cellular level: a) full glucose oxidative phosphorylation; b) anaerobic glycolysis (lactic acid cycle). (Click on the image to enlarge.)
��
Figure 2 Difference in glucose metabolic pathways as a function of oxygen abundance in a) normal cells, and b) cancer cells. (Click on the image to enlarge.) Glycolysis involves the conversion of glucose to pyruvate and then to lactic acid, the waste product. In non-cancerous cells, mitochondria oxidize pyruvate to carbon dioxide and water in the presence of oxygen (Figure 2.a), and the glycolytic reaction is inhibited (Pasteur Effect [[3]]). Conversion of glucose to lactic acid, even in the presence of oxygen is known as aerobic glycolysis (Figure 2.b) or the Warburg effect [[4], [5]]. In one of his seminal papers [[6]], Warburg suggests that carcinogenesis is a two-step process. Cancer cells originate from normal cells by firstly encountering irreversible respiration injury. The second phase of cancer formation represents a long struggle for existence by the injured cells to maintain their structure, in which a part of the cells die from lack of energy while another part succeeds in replacing the irretrievably lost respiration energy by fermentation energy (from lactic acid cycle). Warburg's initial hypothesis that cancer results from impaired mitochondrial metabolism has been shown to be incorrect, but the observation of augmented glycolysis in tumors, even in the presence of oxygen, has been continually proven [[7]]. While cancer cells do carry oxidative phosphorylation, the majority of glucose molecules taken by cancer cells (66%) are metabolized through fermentation [[8]], a process that is ten times faster than full glucose oxidation.
��
Contemporary explanation of the Warburg effect
In addition to being energetically highly inefficient process glycolysis (either anaerobic or aerobic), with its metabolic products (such as hydrogen ions), cause constant acidification of the extracellular space, which might result in increased local toxicity [[9], [10]]. Nevertheless, despite these drawbacks, cancer cells consistently progress towards the wasteful and potentially toxic glycolytic phenotype. Gatenby and Gillies [[11]] proposed that the consistent expression of up-regulated glycolysis is not accidental but represents a solution to the environmental growth constraints during tumor development. They suggest that increased glycolysis is an essential component of the malignant phenotype and, therefore, a hallmark of invasive cancers. Transport enzymes of the Glut and hexokinase families are up-regulated in tumor cells expressing the glycolytic phenotype, and the level of Glut-1 glucose transporter expression has been shown to correlate with [18F] FDG uptake in non-small cell lung cancer, for example [[12]]. Gatenby and Gillies [11] describe the concept of carcinogenesis as a process that occurs by cellular evolution implying that common characteristics of malignant phenotypes, such as upregulation of glycolysis, are the result of active selection processes. They further argue that upregulation of glycolysis is likely to be an adjustment to hypoxia developing as pre-malignant tissue grows gradually further away from their blood supply. Also, an augmented acid production from glycolysis upregulation leads to microenvironmental acidosis and requires further adjustments through somatic evolution to phenotypes resistant to acid-induced toxicity. Finally, they conclude that cell populations that emerge from this evolutionary sequence have a compelling growth advantage, as they alter their environment through increased glycolysis in a way that is toxic to other phenotypes, but harmless to themselves.
��
To further support the attempt by Gatenby and Gillies [11] in explaining the cause of Warburg effect in aggressive tumors as a response to harsh environmental conditions, one might assume that cancer cells ��know�� a priori they will encounter severe conditions in the future. Consequently, they ��decide�� to switch their glucose metabolism to highly inefficient but the only possible (and highly toxic) metabolic pathway. To make their explanation more sounded, Gatenby and Gillies [11] speculate that: ���� intuitively, it would seem that the Darwinian forces prevailing during the somatic evolution of invasive cancers would select against a metabolic phenotype that is more than an order of magnitude less efficient than its competitors and that is environmentally poisonous. In other words, the accepted tenet of ��survival of the fittest�� would seem to generally favour populations with more efficient and sophisticated substrate metabolism.�� Consequently, they suggest that glycolytic phenotype in cancers is directly governed by the evolutive mechanisms, over the relatively short time frame during which tumors develop.
��
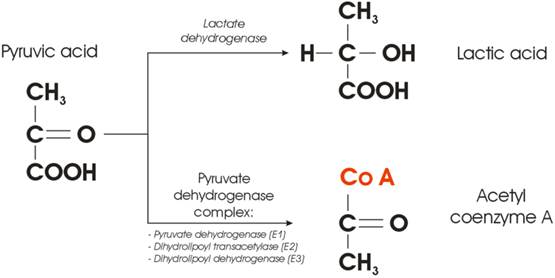
Figure 3 Enzyme-mediated cross-section between lactic and citric acid cycle.
A new hypothesis Figure 3 illustrates the very point within a cellular glucose metabolism where the fate of the Pyruvic acid is decided. In the presence of oxygen, the Pyruvic acid will be converted into acetyl group and attached to coenzyme A, a process mediated by the so-called Pyruvate Dehydrogenase Complex (PDC). The complex consists of three enzymes: pyruvate dehydrogenase, dihydrolipoyl transacetylase, and dihydrolipoyl dehydrogenase. On the other hand, conversion of pyruvic acid to lactic acid requires one enzyme only - lactate dehydrogenase (LDHA). From the cellular kinetics point of view, one may ask a simple question: what is the probability that something goes wrong with either of the two possible metabolic pathways depicted in Figure 3? Synthesis of three enzymes, needed to provide conversion of pyruvic acid to acetyl coenzyme A, even in the presence of oxygen will be (notably three times) more prone to the errors than a transcription of only one (competing) enzyme. It was reported that many human cancers have higher LDHA levels than normal tissues [[13]], but the correlation between oncogenes and glycolysis was poorly understood. The question could be then raised whether the Warburg effect is only a consequence, or could it be at the root of the very cause of carcinogenesis? A new hypothesis could be staged that the switch from aerobic cellular respiration to aerobic glycolysis leads to carcinogenesis, and the cell begins to develop the cancerous phenotype [[14], [15]] at the point where the fate of Pyruvic acid is decided. Such a switch could be governed by the lack of complete PDC (at least one of the enzymes is missing).
��
While at this very moment the question whether Warburg effect is a hen, or an egg remains at the level of pure hypothesis, several clinical observations might be called upon to support such proposition indirectly. While there is no apparent relation between Type 1 diabetes mellitus patients and incidence of cancer to this date [[16]], several publications argue that there could be a lower cancer rate in patients with insulin-dependent diabetes. In 2003, Zendehdel et al. [[17]] published results on cancer incidence in patients with Type 1 (insulin-dependent) diabetes mellitus on a cohort of 29 187 patients, followed over a period of 30 years, during which they observed 355 incidences of cancer. Such a low frequency (1% over 30 years, or 0.04% per year) appears negligible when compared to 1.66 million cases of new cancer cases per year in the US (0.52% per year [[18]]). Pladys et al. [[19]] reported on the lower incidence of cancer death mortality in diabetic patients (6.7%, both Type 1 and 2) when compared to non-diabetic patients (13.4%) using a cohort of 39 811 patients with the end-stage renal disease. It might be argued that cells in diabetic patients (generally deprived of normal glucose uptake due to lacking insulin) become ��trained�� (to use rhetoric by Blagosklonny [[20]]) by the microenvironment and well ��prepared�� as soon as glucose becomes available. Once the glucose is phosphorylated by hexokinase and enters the glucose oxidation process, the cell is ��prepared�� not to waste the opportunity and gets the maximum out of the relatively ��scars glucose supplies.�� One could further argue that diabetic patient cells are making sure that the synthesis of the PDC is up and running flawlessly, to avoid wasteful pathway of cellular glucose metabolism.
On the other hand, despite a relatively small amount of data published it appears that the incidence of cancer is also correlated with the increased intake of carbohydrates [[21], [22], [23]]. One may argue that normal cells, exposed to increased supply of glucose would quickly switch towards the energetically inefficient pathway (lactic acid cycle) of burning glucose even in the presence of oxygen (Warburg effect) since the source of energy (glucose-ATP) are virtually inexhaustible. In addition, the ATP production via fermentation is much faster (as mentioned above), albeit highly ineficient, when compared to full oxydation. It is also of note that contrary to type 1 (insulin-dependent), patients with type 2 diabetes mellitus have higher probability for cancer incidence [[24]]. Yet another detail deserves attention: type 1 diabetes is commonly regarded as a juvenile-onset diabetes as it often begins in childhood while the type 2 diabetes was considered an adult-onset diabetes. However, type 2 diabetes is becoming increasingly common in children [[25]] who are more obese or overweight that could be correlated with carbohydrates rich diets. Finally, the possible triggering of carcinogenesis by aerobic glycolysis, accompanied by increased glucose uptake, can be further supported by studies demonstrating increased glucose uptake observed to coincide with the transition from premalignant lesions to invasive cancer [[26], [27]].
Conclusion
Unlike Warburg's initial hypothesis that cancer cells metabolize glucose through aerobic glycolysis due to impaired mitochondrial function a new hypothesis was presented that the normal cell becomes cancerous at the point when it switches its glucose metabolism from oxidative phosphorylation to aerobic glycolysis. The new hypothesis that Warburg effect corresponds to the very beginning of carcinogenesis might be supported by the eventual failure in synthesis of the PDC. Such failure could be mediated by one (or multiple) of the well-known carcinogenic factors in synergy with an excessive supply of glucose in carbohydrates rich diets. The new hypothesis revolves around a point within cellular glucose oxidation at which the fate of pyruvic acid is decided. Several observations have been presented to support such hypothesis: lower incidence of cancer in insulin-dependent type 1 diabetes mellitus patients; increased cancer incidence in societies consuming high quantities of carbohydrates; and the increased probability of synthesis failure of the pyruvate dehydrogenase complex consisting of three enzymes when compared to the synthesis of a single lactate dehydrogenase enzyme. Contemporary agreement in explaining why tumor cells opt for aerobic glycolysis (Warburg effect) that is far less efficient than oxidative phosphorylation at producing ATP, is that it represents evolutionary adaptation to harsh microenvironmental conditions by using the carbon chains (from the lactic acid) as building blocks for synthesis of biomolecules (nucleic acids, proteins, and lipids), which are essential for cell proliferation. However, even the acceptance of the aerobic glycolysis being a more adequate glucose metabolism pathway for cancer cells, the question of a hen or an egg remains: is the Warburg effect just a consequence, or could it be the very cause of carcinogenesis (Figure 4)?
��

��
Warburg Effect - a Consequence or the Cause of Carcinogenesis?
http://jcancer.org/v07p0817.htm
��
��л�����ڰ�֢�����������еĺ���֢
Metabolism and Its Sequelae in Cancer Evolution and Therapy
Gillies, Robert J. PhD; Gatenby, Robert A. MD
���ء�j������˹��ʿ;���ء��Ǵı�ҽѧ��ʿ
��֢��ͨ��һϵ�п��Ա�����Ϊ��������������¼�����չ�ġ�������Ľ����۵�һ������ǰ���ǣ�������������ѡ�����ʺ��ض����������ֵ�ѹ�������⣬����������(1)����ѡ���ǿ�Ⱥ�(2)��ѡ��Ⱥ�ı��ͷ�������ȡ�ֵ��ע�����,�ڰ�֢�ķ�չ���°����õ���������ת��,ѡ���Ը�ֲ��ϱ仯,����������������,������ϸ����ѡ��,�ı��˴�л����:��ʾ��Щ���ʹ��ڹ�����ѡ�����ƻ�����ѡ��һ������ı����������ǽͽ⣬�������㹻�������ڵ�����������ǵij������͡�������ν�ġ�WarburgЧӦ���Ļ����Ѿ��õ��˺ܺõ��о����ж���ģ�Ϳ��Խ�����������������ڷ���ˮƽ�Ϸ����ġ��ڴˣ������������ͨ�������������ֱ��͵Ļ������������ͳһ�ļ��⡣���仰˵�����ǹ�ע�IJ��ǡ���Ρ����ǡ�Ϊʲô����ϸ�����ֳ��������ǽͽ⡣�������ͨ���������ǽͽ�ĺ���֢������ܴ���һ��ѡ�����ơ����������Ѿ������ǹ��������ϳɴ�л����IJ������ǽͽ�����ں����ķ�Ӧ�ʣ��Ϳ��������IJ���������ǵ���һ������ǣ������ǽͽ�����������;����ϸ����ռ��ữ����ʵ�ϣ��͵�ϸ����pH��ٽ��ֲ���Ϯ���ٽ�ת�ƣ������ƿ��������ߡ�����Ȼ�����İ�֢�У�ϸ����pHֵ������ת�������һ��ǿ����������Ԥ��ָ�ꡣ���⣬���о�����������ϸ�������ж�������ת�ƣ���߿��������ߡ���ˣ�������Ϊ���ڰ�֢����ϸ�����������У���������IJ���Ϊϸ���ṩ��ѡ�������ơ�
Cancers progress through a series of events that can be characterized as
��somatic evolution.�� A central premise of Darwinian evolutionary theory is that
the environment imparts pressure to select for species that are most fit within
that particular microenvironmental context. Furthermore, the rate of evolution
is proportional to both (1) the strength of the environmental selection and (2)
the phenotypic variance of the selected population. It is notable that, during
the progression of cancers from carcinogenesis to local invasion to metastasis,
the selective landscape continuously changes, and throughout this process, there
is increased selection for cells that have altered metabolic phenotypes:
implying that these phenotypes impart a selective advantage during the process
of environmental selection. One of the most prevalent selected phenotypes is
that of aerobic glycolysis, that is, the continued fermentation of glucose even
in the presence of adequate oxygen. The mechanisms of this so-called ��Warburg
effect�� have been well studied, and there are multiple models to explain how
this occurs at the molecular level. Herein, we propose that unifying insights
can be gained by evaluating the environmental context within which this
phenotype arises. In other words, we focus not on the ��how�� but the ��why�� do
cancer cells exhibit high aerobic glycolysis. This is best approached by
examining the sequelae of aerobic glycolysis that may impart a selective
advantage. Many of these have been considered, including generation of anabolic
substrates, response rates of glycolysis vis-��-vis respiration, and generation
of antioxidants. A further sequeala considered here is that aerobic glycolysis
results in a high rate of lactic acid production; resulting in acidification of
the extracellular space. Indeed, it has been shown that a low extracellular pH
promotes local invasion, promotes metastasis, and inhibits antitumor immunity.
In naturally occurring cancers, low extracellular pH is a strong negative
prognostic indicator of metastasis-free survival. Furthermore, it has been shown
that inhibition of extracellular acidosis can inhibit metastasis and promote
antitumor immunity. Hence, we propose that excess acid production confers a
selective advantage for cells during the somatic evolution of cancers.
Metabolism and Its Sequelae in Cancer Evolution and Therapy : The Cancer
Journal
https://journals.lww.com/journalppo/Abstract/2015/03000/Metabolism_and_Its_Sequelae_in_Cancer_Evolution.7.aspx
��
Origin of the Warburg Effect
Project Summary:
Eukaryotic cells have two mechanisms of energy (ATP) production: mitochondrial
respiration and glycolytic fermentation. Through the groundbreaking work of
Louis Pasteur, it was thought that the less efficient fermentation would ensue
only if there was insufficient oxygen to support respiration. This was the
predominant thought until about 100 years ago, when the Nobel Prize-winning
German Physician, Otto Warburg showed that cancer cells ferment glucose at a
high rate, even in the presence of adequate oxygen and sufficient mitochondrial
function (1). The reasons that cancer cells choose to use the less efficient
fermentative pathway over the more efficient respiration have remained a mystery
ever since. We believe that we have solved this mystery, and only need to
convince others. We have argued that as the efficiencies are so different, this
has nothing to do with energy production, where most have been focusing their
time. Instead, we argue, that it must have to do with the production of a
by-product of fermentation (2). We believe that this byproduct is lactic acid,
and have shown through experiments that: 1) acid production is associated with
increased cancer invasion (3, 4), 2) that neutralization of acid can prevent
invasion (5-10), and 3) that stimulation of acid production can promote invasion
(Russell, in prep). Hence, A) fermentation produces acid, B) acid production
improves the fitness and survival of cancer cells that do this, and C) cancers
that ferment are more likely to succeed and be predominant in those that we
detect (11-14).
https://lab.moffitt.org/gillies/research/tumor-ph/origin-of-the-warburg-effect/
References:
Warburg O, Posener K. Uber den stoffwechsel der carcinomzelle. Bioichem Z
1924;152:309-44.
Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose
metabolism of cancers. J Nucl Med 2008;49 Suppl 2:24S-42S.
Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, et al.
Acidity generated by the tumor microenvironment drives local invasion. Cancer
research 2013;73(5):1524-35.
Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ. Acid-mediated tumor
invasion: a multidisciplinary study. Cancer research 2006;66(10):5216-23.
Ibrahim Hashim A, Cornnell HH, Coelho Ribeiro Mde L, Abrahams D, Cunningham J,
Lloyd M, et al. Reduction of metastasis using a non-volatile buffer. Clin Exp
Metastasis 2011;28(8):841-9.
Ibrahim-Hashim A, Abrahams D, Enriquez-Navas PM, Luddy K, Gatenby RA, Gillies
RJ. Tris-base buffer: a promising new inhibitor for cancer progression and
metastasis. Cancer Med 2017.
Ibrahim-Hashim A, Cornnell HH, Abrahams D, Lloyd M, Bui M, Gillies RJ, et al.
Systemic buffers inhibit carcinogenesis in TRAMP mice. J Urol
2012;188(2):624-31.
Ibrahim-Hashim A, Robertson-Tessi M, Enriquez-Navas PM, Damaghi M,
Balagurunathan Y, Wojtkowiak JW, et al. Defining Cancer Subpopulations by
Adaptive Strategies Rather Than Molecular Properties Provides Novel Insights
into Intratumoral Evolution. Cancer research 2017;77(9):2242-54.
Ribeiro MD, Silva AS, Bailey KM, Kumar NB, Sellers TA, Gatenby RA, et al. Buffer
Therapy for Cancer. Journal of nutrition & food sciences 2012;2:6.
Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, et al.
Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer
research 2009;69(6):2260-8.
Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev
Cancer 2004;4(11):891-9.
Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev
Cancer 2008;8(1):56-61.
Gatenby RA, Gillies RJ, Brown JS. Evolutionary dynamics of cancer prevention.
Nat Rev Cancer 2010;10(8):526-7.
Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and
why targeted therapy does not work. Nat Rev Cancer 2012;12(7):487-93.