急性肺损伤的潜在治疗靶点?
缺氧引起的肺部炎症 Hypoxia-Induced Inflammation in the Lung
急性肺损伤的潜在治疗靶点?
图1
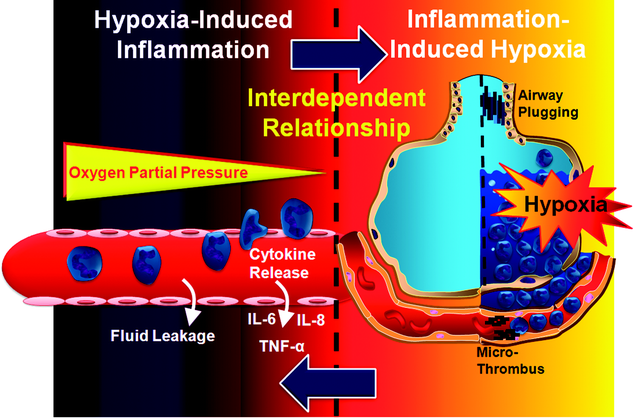
缺氧与炎症相互依赖的关系。暴露在环境缺氧环境下会引发不同器官的急性炎症反应,包括肾脏、肠道、心脏或肺。低氧诱导炎症可以体现为增加血管渗漏,炎症细胞在低氧积累器官,或释放的炎症介质(如肿瘤坏死因子(TNF)
-α,白介素6
(IL),或引发)。与此同时(如急性肺损伤所示),炎症与组织缺氧有关,组织缺氧是由常驻细胞(如肺和血管内皮细胞)和募集的炎症细胞(如中性粒细胞)对代谢物和氧气需求的急剧增加引起的。此外,由于肺水肿、气道阻塞或微血栓,氧供应也会减弱。
急性肺损伤(ALI)是一种严重的低氧性肺疾病,在世界范围内造成大量死亡。尽管最近在支持性护理方面取得了进展,但自从近20年前引入了一个标准的一致定义以来,死亡率并没有明显下降。迫切需要新的策略来帮助设计有效的治疗方法。局部肺泡缺氧是ALI的重要病理特征。由于肺泡缺氧是孤立的,例如在高海拔地区遇到的,在没有任何其他致病性刺激的情况下会导致炎症性肺表型,这些区域可能不是被动的旁观者,但实际上可能有助于肺损伤的发病和进展。肺对缺氧独特的转录反应显然允许它在缺氧水平上表达炎症表型,而其他器官不会产生这种反应。我们将回顾这些独特的转录反应对中度肺泡缺氧的理解的最新进展,这可能为ALI的发病机制提供新的见解。
关键词:ARDS;缺氧;炎症;低氧诱导因子;分子
临床意义
我们对急性肺损伤(ALI)等低氧性肺部疾病的潜在致病机制还不完全了解。肺缺氧的重要促炎作用最近已被认识。这篇翻译综述探讨了在肺中介导这些反应的潜在机制。未来对肺特异性缺氧反应背后的分子机制的阐明可能为我们寻找ALI的治疗策略提供一个新的范式。
考虑到肺泡缺氧是许多呼吸系统疾病不可避免的结果,值得注意的是,缺氧是全身器官强有力的促炎刺激物(1,2)。
肺部疾病中肺泡氧的浓度虽然低于正常值,但却高于引起全身器官缺氧炎症的氧浓度的显著降低。然而,肺泡缺氧导致肺的炎症表型,无论是暴露于低压缺氧的人类,还是暴露于肺泡缺氧的啮齿类动物(与肺部疾病相似)(2,7
- 9)。这些发现表明,缺氧可能在肺中产生破坏性的炎症效应,类似于它对全身器官的影响。因此,肺泡缺氧在肺部疾病中可能不仅仅是一个疾病过程的结果,而是在疾病过程最初建立后可能会积极地促进进行性肺损伤。
然而,考虑到肺缺氧反应发生在一个比系统器官产生缺氧反应的高得多的氧分压(Po2),独特的肺机制一定有助于这种表型。在这篇综述中,我们考虑了支持缺氧作为肺部炎症刺激的作用的最新证据,它发挥这些作用的独特的具体机制,以及这些机制在急性肺损伤(ALI)(一种典型的肺缺氧疾病)发病机制中的潜在作用。
缺氧会导致炎症
缺氧和炎症在分子、细胞和临床水平上相互交织(10)。缺氧(例如由血管阻塞引起)导致器官缺血和促炎表型的表达(11-20)。虽然器官缺血通常发生在无菌环境中,但由此产生的缺氧反应的结果与针对入侵微生物的宿主免疫反应的激活有许多表型相似之处(17)。这种低氧诱导的炎症反应导致免疫细胞的募集、下游信号通路的激活以及促炎细胞因子和趋化因子的诱导(17,18)。此外,这些极低浓度的氧延长了中性粒细胞的生存期,增加了内皮通透性和血管渗漏(11-16)。
除了由缺氧引起外,炎症也是缺氧的一个重要原因。炎症组织由于细胞浸润、水肿和微血栓的形成而变得严重缺氧,这些因素共同延长了毛细血管与代谢活性细胞之间的扩散距离。随后氧传递速率的降低降低了细胞Po2,从而引发炎症,进一步促进了破坏性免疫反应的传播。在类风湿关节炎和炎症性肠病等疾病中,缺氧不是一个旁观者,而是通过调节含氧基因的表达来影响组织的环境,包括促炎网络。导致这一现象的机制目前是许多系统性器官疾病的深入研究的重点,这些研究已经确定了可以在体内模型中进行治疗操作的途径(10)。
调节基因表达以应对缺氧
低氧反应的机制在体内动物模型中已被广泛研究,其中缺氧缺血性条件通常是由血管阻塞引起的,而在体外模型中,氧已减少到0.5-1.0%,模拟缺血时的氧浓度。在这些模型中,几个重要的转录因子参与了基因表达的变化。第一个被确认的是缺氧诱导因子(HIF)家族(11),它对细胞氧浓度的大幅降低做出快速反应,从而构成了缺氧应答基因的关键调节因子(21)。典型的转录因子HIF-1在物种间高度保守,存在于几乎所有的细胞类型中,受到氧气供应的严格调控,并改变了数百个基因的表达。HIF-1存在异质二聚体,由HIF-1α和HIF-1β子单元。HIF-1β既定的存在,而HIF-1α蛋白质在常氧条件下被发现在非常低的浓度。在常氧条件下,由prolyl
HIF-1α的羟基化催化羟化酶域蛋白质(PHDs),使用分子氧作为底物,导致与冯的交互Hipple-Lindau
VHL蛋白和泛素,因此针对HIF-1α蛋白酶体降解(22)。在缺氧,PHDs活动减少,导致HIF-1α稳定,与HIF-1β交互,核易位,DNA结合蛋白,和招募共激活剂蛋白质在缺氧诱导因子结合位点的反应元素,激活靶基因的转录。HIF-2α密切相关的蛋白质,像HIF-1α,受制于PHD-依赖监管和二聚HIF-1β后稳定。然而,HIF-2α细胞表达的展品更受限制的模式(23)。不同的靶基因由HIF的两种亚型控制,对PHDs的不同亚型表现出不同的敏感性(24-27)。
增加HIF-1α蛋白质通常与转录活动增加,尽管HIF-1
transactivation函数进一步受羟基化HIF-1α的天冬酰胺残留通过第二个氧依赖性的羟化酶酶的作用,factor-inhibiting HIF-1
(FIH-1)。在常氧条件下,这种羟基化作用阻碍了转录协同激活因子creb结合蛋白/p300的结合(28)。因为博士和FIH-1活动需要分子氧,缺氧降低了酶的活性,导致HIF-1α的稳定和transactivation
HIF-1的目标基因。
虽然诱导稳定的抑制和激活博士期间和富士康显然是非常重要的快速反应机制的发展深刻的细胞内缺氧,低氧诱导因子也可以激活潜伏期长机制涉及新HIF-α蛋白质的合成(图1)。重要的串扰发生低氧诱导因子和NF-κB之间,和先天免疫系统的激活增加HIF-1αhypoxia-independent的方式通过一个NF-κB-dependent增加HIF-1α基因的转录,数小时或数天的表演(29-36)。另一个低氧独立的HIF蛋白表达增加的机制涉及PI3K/Akt通路对生长因子和细胞因子(包括胰岛素和胰岛素样生长因子)的调节(图2)。该通路还与结肠癌和肺癌中较长期的HIF表达有关(37-40)。另一种有助于HIF稳定的机制最近在缺血性心脏中被描述。低氧诱导增加细胞外腺苷可以导致增加HIF-1α蛋白质通过period-2-dependent机制(41)。
然而,HIF并不是介导缺氧转录反应的唯一转录因子。缺氧对博士的抑制也能稳定HIF以外的蛋白,从而产生不依赖HIF的低氧基因反应(42-46)。促炎转录因子的激活NF-κB在出现缺氧反应,至少在一部分,发生由于博士的抑制。I-κB
kinase-β(IKK-β)包含一个进化为羟基化主题博士守恒的共识。缺氧抑制PHD-1或PHD-2稳定IKK-β并增加其活性,从而导致增加phosphorylation-dependent
I-κB的降解kinase-α(IKK-α)和解放和核易位NF-κB
(42-44)。因此缺氧释放压抑NF-κB活动通过一个HIF-independent分子机制,导致炎症的表现型。有趣的是,ph
-3可以通过不同的hif独立机制介导促炎表型,通过SIVA1/BCL-X(L)依赖途径延长中性粒细胞存活(46)。然而,最近的研究表明,由于先天免疫反应强烈,缺乏PHD3可缩短各种腹腔脓毒症模型小鼠的生存时间,提示在这种情况下,ph
-3具有抗炎作用。相比之下,缺乏pd -1 (pd -1−/)或pd -2 (pd
-2+/−)的小鼠则没有这种表型。因此,博士在调节缺氧诱导的炎症中的作用可能是亚型特异性的,也可能是组织特异性的或器官特异性的(47)。
缺氧还通过诱导microRNA
(miRNA)转录和表达的变化来控制mRNA的浓度。mirna与rna诱导的沉默复合物结合,并在靶mRNA的3’untranslation区域通过互补序列退火,导致翻译抑制和mRNA降解。一组低氧反应的miRNAs(称为“低氧分子”)控制低氧细胞中的mRNA浓度,从而改变表型反应。hypoxamirs本身的转录是由hypoxia-responsive转录因子,因为microrna是嵌入区域的基因的转录受这样的转录因子,包括低氧诱导因子和NF-κB(48岁,49)。有趣的是,据报道,通过翻译后对mirna结合蛋白argonaute2
(Ago2)的修饰,hypoxamirs的作用也发生了转录独立的改变(50)。型胶原proyly-4
-羟化酶(C-P4H9(I))羟化Ago2,从而在缺氧开始后4小时内激活并稳定蛋白。这种羟化作用增加了大量mirna的表达,增加了Ago2的内切酶活性。C-P4H9(I)是一种prolyl羟化酶,它不同于已经讨论过的1-3位博士,并且其对氧的Km值要低得多,因此在深度缺氧环境中仍然保持活性(50)。此外,缺氧时C-P4H9(I)的表达通过一种与hif无关的蛋白稳定机制增加(51)。
综上所述,这些数据表明,极低水平的缺氧,如缺血器官中遇到的缺氧,会抑制prolyl羟化酶,导致基因转录和mRNA的稳定性发生改变,尽管其机制既依赖于hif,也不依赖于hif。
肺对缺氧的特异性反应
缺氧的经典定义是“不能提供足够的氧气来维持组织ATP的供应”,但这在肺中通常是不适用的,因为肺中的缺氧反应发生在氧气浓度完全足以维持正常的氧化代谢的情况下。正常肺泡Po2(约110毫米汞柱)与其他内部器官相比是唯一的高,在健康状态下,氧张力范围为3-20毫米汞柱(52-55)。此外,低氧反应是观察肺在Po2值(50
- 60毫米汞柱)高于其他器官的正常条件下氧化,并明显大于在其他任何器官缺血性和炎症条件下产生低氧反应的(Po2值< 10毫米汞柱)(52,56,57)。
除了独特的高Po2水平,与其他器官相比,肺对缺氧有独特的反应。两个具体的回答清楚地说明了这一点。首先,急性缺氧导致血管收缩(缺氧性肺血管收缩),即与其他所有器官缺氧引起的血管舒张形成明显对比(58,59)。这是一个非常快的(即。当疾病局限于肺内少数区域时,这是一种有益的适应,因为它改善了呼吸-灌注匹配,提高了吸氧能力(58)。然而,在一些正常的海平面居民(占人口的5-10%)中,过度的血管收缩反应与高海拔肺水肿的发展有关(60)。更持久的肺部缺氧(持续数周甚至数月或更长)引起肺动脉高压引起肺小动脉重塑和持续ρ-kinase-dependent血管收缩剂反应,不同于急性缺氧性血管收缩(61、62)。在高海拔地区,与任何潜在的肺部疾病隔离,这种高血压可导致右心衰和易感人群的死亡,如亚急性山地病和慢性山地病(蒙格病)(60,63)。
肺对缺氧反应的第二个独特方面涉及特定基因,这些基因的转录在肺中被改变,但在其他器官中不被激活。邓和他的同事报道,抵抗素类似物-α(RELM-α)上调肺缺氧的模仿那些肺部疾病,但并不是在任何其他器官(64)。此外,RELM-α施加促炎效应通过血管内皮生长因子受体2
(VEGFR2)端依赖和IL-4-dependent通路。此外,RELM-α 促进血管生成,在啮齿动物RELM-α
的过度表达导致肺动脉高压的发展,肺血管阻力增加,肺小动脉的重建(65 - 67)。相反,RELM-αsiRNA-调节敲除减缓缺氧导致的肺动脉高压的发展(66)。
利用一种独特的减法基因芯片技术,我们发现了一组在肺缺氧微血管内皮中表达上调的基因,其在全身微血管缺氧内皮中的表达没有改变或减少。到目前为止,对其中两个基因进行了更广泛的研究,即gremlin1和CXC趋化因子受体7型(CXCR7)(68)。两者在缺氧肺中均上调,但在其他(全身)器官中未发生改变(68,69)。gremlin1的单倍体缺乏增强了低氧小鼠肺中的骨形态发生蛋白信号,并通过减轻肺血管重构降低肺血管阻力,从而证明gremlin在低氧性肺动脉高压的发展中具有重要的机制作用(69)。重要的是,gremlin1在移植肺的血管内皮中表达增加(69)。
我们在缺氧小鼠肺中发现的第二个选择性上调的基因是CXCR7,这是一种趋化因子受体,它结合两个配体,趋化因子(C-X-C
motif)配体(CXCL)11和CXCL12(70)。CXCL12是中性粒细胞、t淋巴细胞和单核细胞的趋化剂,促进移行细胞的迁移,并发挥强大的促血管生成作用(71-75)。我们最近报道了CXCR7在人肺高压肺中的表达增加,其配体CXCL12在人肺高压中显著升高(76)。CXCR7的阻断可抑制小鼠肺内低氧性肺动脉高压的发展,提示该受体具有重要的机制作用(77,78)。综上所述,这些报道表明肺选择性缺氧反应基因发挥促炎和致病性血管作用,可能在肺缺氧疾病的发展中发挥重要作用。
除了这些lung-specific基因对缺氧的反应,其他缺氧反应发生肺不是独一无二的,但这引发了氧气的浓度远远高于那些需要系统性的器官,如炎症、血管生成和表达内皮细胞的促凝血的表现型(61、79、80)。中等水平的肺泡缺氧,如暴露在高海拔环境中,可导致炎症表型。增加巨噬细胞、中性粒细胞和炎性细胞因子,包括IL-1β,il
- 6,引发,TNF-α,在支气管肺泡灌洗液的人类暴露于低比重的缺氧(81)。同样,暴露于肺泡缺氧环境的啮齿类动物(FiO2 =
0.10)表现出肺中性粒细胞浸润、巨噬细胞募集和活化、肺血管通透性增加、肺内炎性细胞因子升高(2,7 -
9)。这些发现表明,肺缺氧(尽管Po2水平高得多)可能会产生与全身器官类似的破坏性炎症效应。此外,除了局部肺泡炎症外,肺缺氧还会诱导活化的肺泡巨噬细胞释放单核细胞趋化蛋白-
1,然后将其释放到循环中,在肺泡缺氧开始的几分钟内刺激全身器官的炎症反应(82-84)。因此,肺对缺氧的特异性反应可以引起重要的全身效应。
除了促炎表型的表达外,中度肺泡缺氧刺激肺循环中的血管生成。然而,在新血管形成的过程中,毛细血管内皮出现渗漏,可能导致急性缺氧肺肺泡水肿的形成(61,79)。新血管形成是类风湿关节炎和炎症性肠病炎症过程中的重要致病机制,新血管形成也可能加重炎症性肺部疾病(85)。
肺特异性缺氧反应的机制
考虑到缺氧肺中激活的独特的基因网络和这些反应发生时独特的高Po2水平,肺特异性机制必须控制肺对缺氧的炎症反应。缺氧后迅速激活的转录因子可能在启动基因和蛋白表达的变化中起关键作用。三个转录因子,诱导,环腺苷酸(营)反应元件结合蛋白(分子)和NF-κB(86
- 88),证明这种早期反应在其他器官缺氧,而且每个将被认为是在这里。
低氧诱导因子(HIF)
部分缺乏HIF-1α表达(HIF-1α+ /−)杂合的老鼠暴露在缺氧(供给= 0.1),但在延长缺氧暴露(>
3周)实现右心室肥大的发展,肺动脉高压,和肺血管重建的部分短期保护,这些反应与他们的野生型的同胞是一样的(86)。然而,HIF-2α-缺少 (HIF-2α+
/−)动物缺氧导致肺动脉高压的获得长期保护(87)。HIF在低氧肺反应中的重要性进一步通过观察Chuvash病患者得到证实。在这种情况下,降解率HIF-1α和HIF-2α减少由于VHL基因的突变,导致持续的低氧诱导因子浓度增加。患有这种遗传性疾病的患者表现出严重的肺缺氧反应,并且由于一生中暴露在高浓度的HIF环境中,不可避免地发展为肺动脉高压(89)。复制了Chuvash病基因突变的小鼠也出现了自发性肺动脉高压的发展。当与小鼠缺乏HIF-2α(HIF-2α+
/−)交叉,这些老鼠对肺动脉高压保护,虽然他们HIF-1α+ /−老鼠交叉时不获得保护,提供HIF-2α对肺对缺氧反应的关键作用的进一步的证据(90)。
HIF-1α-缺少和HIF-2α-缺少老鼠和楚瓦什人( Chuvash)综合症患者获得的证据清楚地表明,肺对缺氧的反应持续数周或数年,涉及低氧诱导因子通路。然而,这些变化是否在这些事件中扮演了早期的主要角色还不清楚。在体外培养的细胞模型中,使用肺内未遇到的氧浓度(0.5-1.0%),通常证明了PHD对HIF的稳定作用。Yu和他的同事证明了,在孤立通风的雪貂肺缺氧诱导HIF-1α迅速,早期的增加,但只有当肺泡氧气减少1.3%(∼9毫米汞柱)时。在完整的生物体中,这样的肺泡氧浓度与存活数分钟不相容(91)。因此,尽管HIF对较长期的肺缺氧反应无疑是重要的,但快速、依赖于PHD的HIF稳定不太可能是体内缺氧肺中基因表达早期改变的原因。
cAMP反应元件结合蛋白
普遍存在的转录因子CREB通过丝氨酸133的磷酸化被多种机制激活,包括蛋白激酶a的典型的camp依赖性激活(图1)。这种磷酸化导致多个基因的表达被调节(92)。
我们最近报道了一系列的调查进行识别转录因子通路和基因选择性地激活肺泡肺反应水平的缺氧中遇到人类的肺部疾病,我们发现分子的转录因子家族成员(CREB-1和激活转录因子-
1)选择性地激活磷酸化在缺氧性肺在3小时的接触,但不是在其他器官(88)。四CREB-dependent基因已经被测试日期(脑源性利钠因子,endothelin-1――卵泡抑素――过多,VEGFR-1),证明是上调和肺部缺氧,但一成不变的全身器官,从而提供进一步支持的一个重要的角色lung-selective分子活化途径应对缺氧(88)。
Chava和他的同事(93)最近证实了CREB在维持肺血管功能方面的关键作用,无论是在基础条件下还是在对炎症介质的反应中。在小鼠lps损伤的肺中,显性阴性CREB突变体的表达破坏了CREB的功能,增加了基础肺微血管的通透性,阻止了肺水肿的消退(93)。
因此,CREB在肺泡缺氧时被迅速激活,这与肺部疾病中发现的情况类似,可能在肺特异性缺氧反应中发挥重要作用。
NF-κB
NF-κB组成一个家庭通常激活的转录因子和促炎细胞的刺激后配体,但是已经讨论过的,也可以被激活,以应对缺氧(94、95)。类似于分子,NF-κB是“迅速行动”的主要转录因子结构上存在于细胞溶质的不活跃的形式。激活信号使其离解的抑制蛋白质I-κB及其快速核易位,调节基因的功能(图2)。
NF-κB迅速被激活(2小时内)在肺缺氧反应(9)体内,在阿里中常见的肺泡氧气浓度(10%)、转基因小鼠表达荧光素酶NF-κB证明缺氧的控制下选择性地激活NF-κB肺部,导致cyclooxygenase-2和其他重要的促炎基因的转录激活(94)。Sarada和同事们发现一个健壮的核NF-κB增加肺的啮齿动物暴露于低比重的缺氧和显著增加NF-κB
dna结合活性(96)。此外,封锁NF-κB,使用多酚化合物姜黄素在缺氧暴露之前,导致核NF-κB浓度,减少,相应的肺水含量和transvascular泄漏(96)。
然而,在肺NF-κB激活的机制还有待充分阐明。虽然被激活以响应通过PHD-dependent机制系统性器官缺氧,博士学位不太可能负责的活动NF-κB或肺的易位,鉴于独特高警察乙。为了更全面地了解它在肺中的作用,需要确定和表征肺泡水平Po2的激活机制。尽管如此,考虑到这是(1)激活早期在缺氧反应的氧浓度符合那些在阿里,(2)控制炎性细胞因子网络,和(3)维护重要的相声低氧诱导因子通路NF-κB可能发挥重要作用的早期肺缺氧反应的起始和传播。
最近,许多出版物记录了低氧诱导的低氧肺中特异性microrna的增加,并表明阻断它们的活动可以预防肺动脉高压的发生(50,97 -
101)。然而,到目前为止,还没有发现这些miRNA表现出早期的肺选择性激活,也没有发现任何控制miRNA转录和表达的肺特异性机制。
缺氧可能是Ali的发病机制之一
ALI是一种典型的低氧肺疾病,表现为进行性动脉低氧血症、呼吸困难和呼吸功显著增加。虽然有几个不同的初始事件与它的发展有关,但所有的结果都是相似的临床和病理特征,包括局部肺泡缺氧和严重的动脉低氧血症的发展(102)。
利用多重惰性气体排泄技术获得的数据表明,ALI肺区域的范围从高或正常的通气灌注比到低的通气灌注比和不通气区域(V./Q.)。<
0.005,也就是说。肺内的分流器)。根据疾病严重程度(103-107),流向低通气区和肺内分流区的血流量分别占心脏输出量的5-38%和15-63%。在肺通气不良的区域,Po2的值可以从接近平均肺泡Po2的值到类似于混合静脉血的值(∼32mm
Hg In ALI)(108)。因此,在这些低v /Q中。大比例肺泡暴露于与发生肺特异性缺氧反应相适应的氧张力水平。
有趣的是,现代肺保护策略和应用更高水平的呼气末正压(PEEP)并没有减少ALI患者不通气的肺组织区域。Feihl和同事发现,在一组按照ARDSnet指南进行通风的人群中,包括低潮气量允许性高碳酸血症和中等水平的PEEP,与传统的高潮气量通气策略相比,分流率(48%±5%)增加(105)。因此,尽管采用了现代的肺保护通气技术,但ALI中多达一半的肺暴露在缺氧环境中,仅靠机械通气或补充氧气无法克服缺氧。大比例肺泡及其邻近的肺血管内皮暴露于已知能产生低氧炎症反应的氧浓度下,增加了这些区域在ALI进展中起重要作用的可能性。这些区域的低氧炎症反应一旦开始,可能会促进一种自我持续的放大级联反应,从而使低氧加重炎症性肺损伤,进而导致肺泡进一步缺氧(图3)。
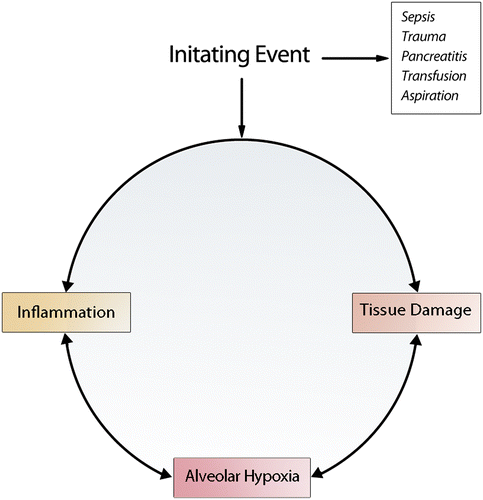
图3。
多种损伤可导致肺损伤的发展。无论初始的损伤是什么,组织损伤的区域都是不均匀的,导致局部肺泡缺氧。损伤性缺氧诱导的炎症反应在这些区域的激活导致了一种自我持续的放大级联,从而使组织损伤恶化,导致肺泡进一步缺氧。
肺动脉高压是急性肺损伤的一个重要并发症,其重要性在最近的研究中得到了强调,肺血管阻力的增加是不良预后的一个独立预测因子(109)。由于肺泡缺氧,在没有任何其他刺激的情况下,可导致肺动脉高压、右心衰和易感人群的死亡(如亚急性高原病),它可能是严重ALI病例中血管病理学的主要贡献者(60,63)。综上所述,这些数据表明,对肺特异性缺氧反应机制的进一步了解可能增强我们对ALI致病机制的了解,并可能揭示该疾病新的治疗靶点。此外,这些反应的肺选择性可能提供了对机制的深入了解,以最小的不必要的系统性影响为目标。
结论
我们对ALI等低氧性肺部疾病的潜在致病机制的认识还不全面。肺缺氧的重要促炎作用最近被认识到,并且已经开始研究介导这些反应的器官特异性机制。未来说明HIF-1的不同功能的肺、分子机制的探索缺氧NF-κB激活,并理解pulmonary-specific激活和抗炎作用的分子可能在我们的搜索提供了一种新的范例缺氧性肺疾病的治疗策略,包括阿里。
Hypoxia-Induced Inflammation in the Lung | A Potential Therapeutic Target in
Acute Lung Injury? | American Journal of Respiratory Cell and Molecular Biology
https://www.atsjournals.org/doi/full/10.1165/rcmb.2012-0137TR
针对低氧诱导的炎症 Targeting Hypoxia-induced Inflammation
在最新一期的《麻醉学》杂志上,Kim等人发表了一篇来自医学博士H. Thomas
Lee研究实验室的文章通过急性肾脏损伤(AKI)的实验模型,研究抑制肾脏缺血引起的炎症和随后的多器官衰竭的策略。事实上,手术患者发病和死亡的主要原因之一是围手术期器官衰竭,如AKI、肝功能衰竭、肠或心肌缺血、中风或急性肺损伤。这些围手术期疾病的一个共同特点是存在缺氧诱导的炎症。2、3在这些情况下,缺氧与炎症的关系是相互依赖的。例如,在高海拔登山时暴露在环境缺氧环境中,会导致肺部或脑部水肿以及人体的全身炎症反应。类似地,短期暴露于环境缺氧(例如,4
-
8小时内8%的氧气)会导致炎性细胞因子浓度升高和肺水肿。此外,在器官移植过程中,供体器官长时间暴露在缺血环境中会增强移植物炎症和早期移植物衰竭。以内毒素受体toll样受体4
(endotoxin receptor toll-like receptor 4,
TLR4)为例,在肾移植过程中,供体肾脏中有TLR4表达,且随着缺血时间的延长,TLR4表达浓度增加。带有TLR4受体功能缺失突变的供体肾脏显示出肾脏炎症减弱和即刻移植物功能增加。综上所述,这些研究表明缺氧是一种炎症刺激(图1),提示靶向缺氧诱导的炎症可能是围手术期药物治疗的重要途径。
图1所示。缺氧与炎症相互依赖的关系。暴露在环境缺氧环境下会引发不同器官的急性炎症反应,包括肾脏、肠道、心脏或肺。低氧诱导炎症可以体现为增加血管渗漏,炎症细胞在低氧积累器官,或释放的炎症介质(如肿瘤坏死因子(TNF)
-α,白介素6
(IL),或引发)。与此同时(如急性肺损伤所示),炎症与组织缺氧有关,组织缺氧是由常驻细胞(如肺和血管内皮细胞)和募集的炎症细胞(如中性粒细胞)对代谢物和氧气需求的急剧增加引起的。此外,由于肺水肿、气道阻塞或微血栓,氧供应也会减弱。
缺氧与炎症相互依赖的关系。暴露在环境缺氧环境下会引发不同器官的急性炎症反应,包括肾脏、肠道、心脏或肺。低氧诱导的炎症可表现为血管渗漏增加、缺氧器官炎症细胞积聚或炎症介质释放(如
、肿瘤坏死因子(TNF) -α白介素(IL)
6,或引发)。与此同时(如急性肺损伤所示),炎症与组织缺氧有关,组织缺氧是由常驻细胞对代谢物和氧气的需求急剧增加引起的。
、肺和血管内皮)和募集的炎性细胞(如
中性粒细胞)。此外,由于肺水肿、气道阻塞或微血栓,氧供应也会减弱。
虽然缺氧可以引发炎症反应,但炎症本身是组织缺氧的一个原因。在活动性炎症期间,代谢向缺氧的转变是严重的。以严重组织缺氧为特征的炎性疾病的一个例子是急性肺损伤。在这种情况下,有几个因素会导致肺缺氧,包括由于气道不张导致的氧供应减少和流向肺内通风区域的血液减少。此外,常驻细胞或募集的炎性细胞增加代谢需求,导致代谢物和氧的供需比发生深刻变化(图1),缺氧驱动的信号通路被激活。9、10组织缺氧在炎症过程中不仅仅是简单的旁观者效应;通过调节氧依赖基因的表达,可以极大地影响炎症的发生或减弱。事实上,针对转录调节组织适应缺氧的研究是防止缺氧引起的炎症和器官衰竭的一个重点研究领域。例如,针对缺氧驱动的炎症的实验策略已被提出用于肠、心肌或肝脏缺血的治疗;急性肺损伤;或AKI.12
AKI.13
AKI的特点是肾小球滤过率下降,发生在几分钟到几天内。在住院患者中,超过50%的AKI病例与肾缺血有关,或超过80%与危重监护有关。最近一项对住院病人的研究表明,只有血清肌酐浓度(0.3-0.4
mg/dl)的轻度升高,其死亡风险比未升高的人高出70%。此外,需要交叉夹持主动脉和肾血管的外科手术与高达30%的肾衰竭率相关。同样,在正常情况下,超过10%的患者在心脏手术后发生AKI,并与死亡率的急剧上升有关。此外,AKI和慢性肾脏疾病是肝移植术后常见的并发症。例如,肝移植后AKI的发生率至少为50%,8-17%的患者需要肾脏替代治疗。在肾移植过程中,移植功能的延迟常常与缺血相关的急性肾损伤有关
正如Kim等人目前的研究显示,1个缺血引起的AKI触发一个向下的螺旋,导致随后的多器官衰竭。作者优雅地论证了暴露于肾缺血并伴有肾脏炎症激活将导致随后的肠道损伤,包括肠上皮屏障的破坏和大量的肠道炎症。肠道损伤最终会扩散到肝脏,引起严重的肝脏疾病。1令人意外的是,这种恶性循环是由局部损伤肾脏(图2),因此,这将是至关重要的理解相声机械方面的通路连接肾脏和肠道在hypoxia-driven肾脏炎症和确定肠道炎症触发后续肝损伤和multiorgan失败。目前的研究表明,挥发性麻醉药可抑制AKI.1引发的肾脏炎症和多器官功能衰竭。通过结合药理学研究和鞘氨醇激酶抑制剂,以及基因方法敲除小鼠的鞘氨醇激酶,他们优雅地证明了这一途径在肾脏保护中的功能作用。这与他们之前的研究一致,20
-
22显示通过鞘氨激酶和鞘氨醇-1-磷酸途径保护肾脏。有趣的是,李氏实验室的其他实验研究表明,局部麻醉输注(如利多卡因、布比卡因或丁卡因)可增强缺血再灌注损伤后的肾功能障碍和肾脏炎症,从而起到相反的作用
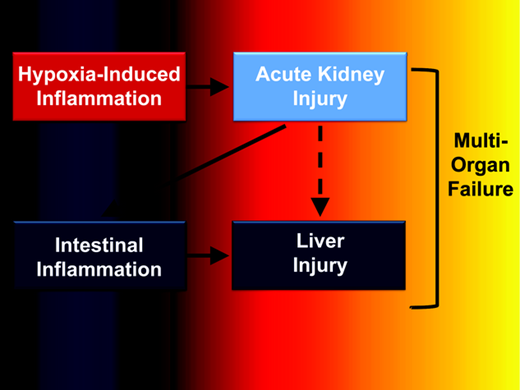
图2所示。在急性肾损伤引起的多器官功能衰竭中所提出的串音通路(参见Kim等人1)。
目前的研究与Lee的团队先前的研究一致,证明了细胞外腺苷信号在AKI中的保护作用。在细胞外腔室中,腺苷来源于前体核苷酸的磷酸水解,如三磷酸腺苷和一磷酸腺苷。事实上,在缺氧的情况下(例如,在AKI期间发生的情况),细胞外腺苷的产生会显著增加。24-26细胞外腺苷的产生和信号转导与低氧性炎症的减弱密切相关。因此,当暴露于肾缺血时,细胞外腺苷酶生成缺陷的转基因小鼠更容易发生肾脏炎症和器官功能障碍。27,28胞外腺苷可通过四个单独的腺苷受体(ARs)发出信号:A1AR、A2AAR、A2BAR和A3AR。在此背景下,Lee的研究团队的几项研究已经证明了A1AR在减轻肾脏炎症和保护缺血时器官功能方面的重要作用。例如,A1AR基因缺失的小鼠在缺血再灌注损伤后肾脏损伤增加。此外,当A1AR−/−小鼠暴露于感染性腹膜炎时,其死亡率、肾功能障碍和肝损伤均有所增加。此外,一项使用病毒过表达技术的优雅的原理证明研究31表明,A1AR−/−小鼠肾特异性A1AR重建可减少肾缺血再灌注损伤。目前的研究显示肾脏、肠道和肝脏之间存在着一种串音通路,与此一致的是,1
Lee的研究团队能够证明通过激活a1ar介导的Akt来保护肝脏免受AKI的损害。其他32项研究表明,低氧诱导的A2BAR可以保护肾脏免受缺血。综上所述,这些研究强调了由缺氧依赖信号通路激活的内源性抗炎通路可以靶向治疗缺血引起的肾脏炎症。
尽管许多实验研究,包括Lee的研究团队令人兴奋的贡献,为预防或治疗由低氧引起的肾脏炎症提供了新的见解,但将这些方法应用于临床仍然是一个挑战。事实上,在围手术期预防或治疗AKI的治疗方法极其有限。例如,一些涉及多种药物的临床试验显示出很少或没有保护作用。调查多巴胺在围手术期预防急性肾损伤中的作用的研究表明,多巴胺并没有降低死亡率或肾脏预后。此外,研究34调查了作为肾脏保护的一种手段的循环利尿剂(如速尿),没有发现降低整体死亡率。同样,关于选择性d1
-多巴胺受体激动剂非诺多巴胺的保护作用的研究也未能证明其在围手术期肾脏保护的常规临床应用是合理的。因此,未来的挑战将包括系统的方法来帮助将肾脏保护的新实验方法从实验室转化到临床,从小鼠转化到人类。此外,对抑制低氧诱导的肾脏炎症的转录调控通路的进一步机制研究,有望有效治疗低氧诱导的炎症。通过研究组织适应缺氧的内源性机制,这些研究可以解决由微rna调控的肾基因表达的缺氧依赖性变化或缺氧感知和信号机制在肾脏疾病中的直接作用。事实上,我希望这样的研究能让我们进一步开发出治疗策略,在降低缺氧引起的炎症的同时增加缺血的抵抗力。这些治疗方法可以应用于许多围手术期的情况,包括缺氧引起的器官功能障碍,包括急性肺损伤,器官缺血,或实体器官移植。
黏膜炎症项目,麻醉科,科罗拉多大学丹佛,奥罗拉,科罗拉多。
Targeting Hypoxia-induced Inflammation | Anesthesiology | ASA Publications
https://anesthesiology.pubs.asahq.org/article.aspx?articleid=1933201