线粒体活性氧和癌症 Mitochondrial Reactive Oxygen Species and Cancer
摘要
线粒体产生活性氧(mROS)是电子传递链活动的自然副产品。虽然最初的研究集中在活性氧的破坏作用上,但最近的研究表明,mROS可以作为信号分子激活促生长反应。长期以来,人们一直观察到癌细胞相对于正常细胞产生更多活性氧,尽管这种增加的影响并不总是很清楚。考虑到癌细胞通常也会诱导抗氧化蛋白的表达,这一点尤其有趣。在这里,我们讨论癌症相关的突变和微环境如何增加mROS的产生,从而导致致瘤信号的激活和代谢重编程。这种致瘤信号也增加了抗氧化蛋白的表达,以平衡活性氧的高产量,维持氧化还原的稳态。我们还讨论了癌症特异性的活性氧和抗氧化剂的修饰如何成为治疗的目标。
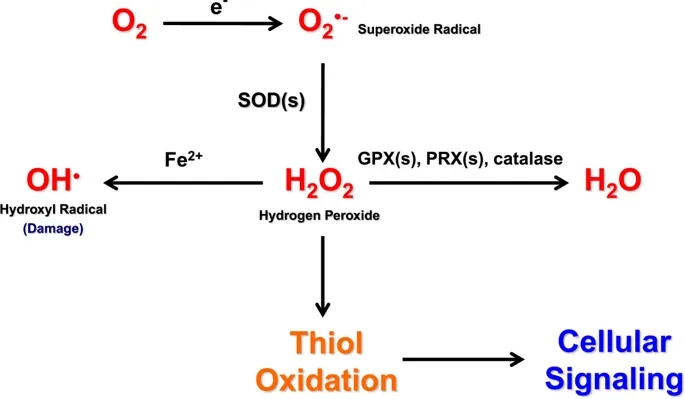
活性氧的产生和相互转化。O2・−是通过从NADPH氧化酶(NOX)酶或线粒体电子传递链中的电子泄漏中获得一个电子而形成的分子O2。超氧化物歧化酶(SOD)酶将两个超氧化物分子转化为一个H2O2和一个水(H2O)分子。过氧化氢可与Fe2+发生芬顿化学反应生成HO・,该反应活性极高,可引起细胞损伤。过氧化氢也可以修饰氧化还原敏感的半胱氨酸残基来改变细胞信号。另外,过氧化氢可以通过谷胱甘肽过氧化物酶(GPXs)、过氧化物酶(PRXs)或过氧化氢酶还原为水。
活性氧的来源
细胞内ROS的一个主要来源是NADPH氧化酶。NADPH氧化酶催化O2和NADPH生成超氧化物。这些酶最初是在吞噬细胞中被发现的,在吞噬细胞中,它们通过在局部产生高水平的氧化应激[9]来杀死被吞噬的病原体。自这一发现以来,已经观察到NADPH氧化酶家族成员存在于机体的许多组织中,这些组织对非免疫功能也很重要[10,11]。专门产生活性氧的酶的存在证实了活性氧在细胞中起控制作用的模型,而不是简单地充当有毒的副产品。此外,致癌基因可以刺激NADPH氧化酶依赖的ROS产生,这已被证明是细胞增殖[12]所必需的。NADPH氧化酶在细胞内定位于许多细胞器,包括质膜、细胞核、线粒体和内质网。有趣的是,内质网最近也被证明有NADPH氧化酶独立产生ROS和[13]。虽然NADPH氧化酶是细胞内ROS的良好来源,但如果可能,这篇综述将着重于线粒体来源的ROS的机制和后果。
细胞ROS的最大贡献者是线粒体。据估计,线粒体总耗氧量的1%用于产生超氧化物[14,15]。线粒体有八个已知的能够产生超氧化物的位点[16,17]。这些位点对细胞总ROS的相对贡献尚不清楚,然而,来自复合物I、II和III的ROS都被证明对细胞信号转导[16]有影响。有趣的是,复合物I和复合物II在线粒体基质中释放ROS,复合物III则有能力在线粒体内膜[18]两侧释放ROS。从理论上讲,将活性氧释放到内膜空间将更容易接近胞质靶。与这一假设相一致的是,复杂iii衍生的ROS已被明确证明在许多生物学过程中需要,包括氧感应、细胞分化和适应性免疫[19]。mROS的其他来源是否对整个mROS信号转导起单独或共同作用尚不清楚。
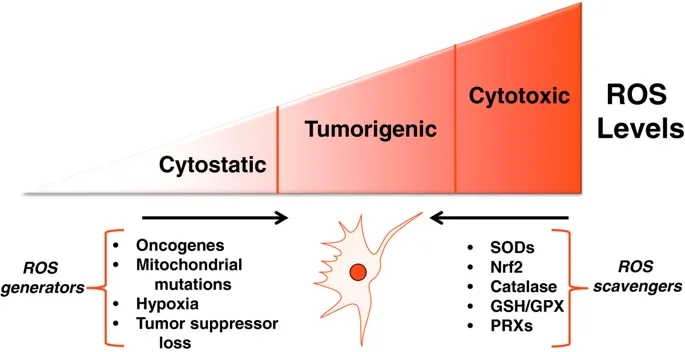
平衡活性氧生成和活性氧清除使癌细胞保持在活性氧水平的致瘤范围内。通过癌基因、线粒体突变、缺氧或肿瘤抑制因子丢失激活线粒体ROS生成,增加ROS信号以增加致瘤性。肿瘤细胞也表现出抗氧化蛋白水平的增强,这些抗氧化蛋白可阻止增加的活性氧达到与生长不相容的细胞毒性水平。
ROS增强磷酸肌醇3-激酶信号(PI3K)
PI3K信号通路是一种中枢生长因子反应通路,在许多癌症中高度激活。激活这一途径已被证明可以增加增殖,促进生存,并增加细胞移动性[34]。

活性氧修饰细胞信号。来自NOXs或线粒体的过氧化氢可以激活PI3K通路、缺氧诱导因子(HIF)通路和代谢适应。这些修饰对肿瘤的生存、生长和增殖至关重要。
细胞内ROS水平可影响PI3K通路。外源性过氧化氢处理细胞足以激活Akt[35]。PI3K通路中已知的主要ROS靶点是PTEN。已有研究表明,ROS可氧化PTEN
(Cys124)上的活性位点半胱氨酸,导致二硫形成形成另一种蛋白内半胱氨酸(Cys71)。这导致PTEN失活,PI3K通路永久活化[36,37]。除了一般的ROS作用外,mROS还可以抑制PTEN并激活Akt[38,39]。除了PTEN,
ROS还被证明可以抑制其他磷酸酶,包括蛋白磷酸酶2A (PP2A)和蛋白酪氨酸磷酸酶1B
(PTP1B)[40]。PP2A去磷酸化Akt的苏氨酸308和丝氨酸493,导致Akt失活;然而,PP2A的去磷酸化活性被双氧水[41]所抑制。PTP1B也通过去磷酸化抑制Akt活性,但与PP2A一样,ROS抑制PTP1B活性并增加Akt活性,从而增加了锚定无关生长[42,43]。因此,ROS抑制磷酸酶,使PI3K信号通路失调,导致Akt信号通路增加,促进增殖和存活(图3)。
线粒体ROS激活缺氧诱导因子(HIF)
对mROS反应的最佳特征通路之一是缺氧反应通路。缺氧是体内肿瘤细胞的一个显著特征,因为肿瘤细胞的高增殖率与血液供应提供包括氧气在内的营养物质的能力不匹配。肿瘤细胞激活缺氧诱导因子(HIFs)激活转录网络,使肿瘤细胞适应缺氧微环境。通路包含三个hypoxia-sensitiveα亚基(HIF1α、HIF2αHIF3α),激活后,与既定的二聚化HIF1β表达和激活转录从hypoxia-response元素(人力资源)[44]。在常氧条件下(21%
O2) HIFα子单元正在迅速对脯氨酸残基的羟化prolyl羟化酶domain-containing蛋白2
(PHD2)认可和针对退化的冯Hippel-Lindau (VHL) E3泛素连接酶途径[45]。当细胞处于缺氧,PHD2
HIFα亚基的羟基化是抑制导致HIFα积累,heterodimerization,核易位。HIF异源二聚体与共激活因子p300和CBP相互作用,启动HREs低氧反应基因的转录。对于缺氧条件下的细胞,HIFs的转录靶点包括促进缺氧条件下存活的基因、将代谢转化为糖酵解增加的基因以及激活血管生成的[46]基因。
暴露于低氧增加mro稳定HIFα子单元。初始证据这种机制源于观察细胞耗尽他们的线粒体DNA(ρ0细胞)不能稳定HIFα子单元在缺氧[47]。
因此,mro都足够和缺氧激活所需的低氧诱导因子(图3)。有趣的是,抑制HIF1α的抗氧化剂治疗已被证明在体外抑制肿瘤细胞增殖和体内[58 59]。
癌细胞增加线粒体活性氧
致瘤突变增加mROS
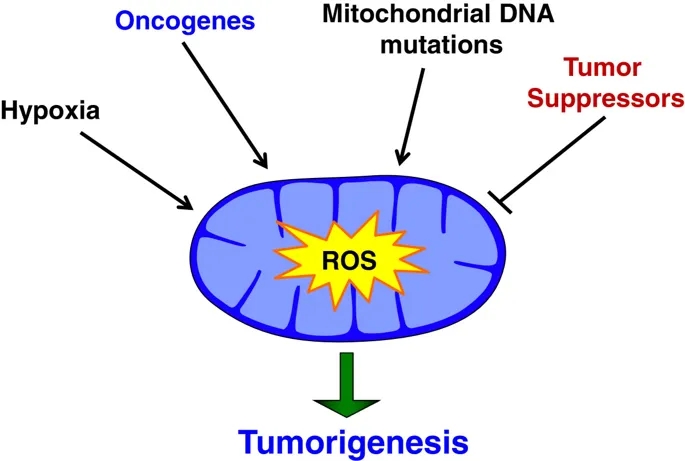
许多癌细胞的活性氧水平升高,增加活性氧的信号事件和突变是一个活跃的研究领域。有几个致癌基因与ROS生成增加有关(图4)。H-RasG12V的外源性表达已被证明可增加3T3成纤维细胞的有丝分裂活性,而该活性依赖于ROS[12]的增加。在显性p53阴性的小鼠胚胎成纤维细胞(MEFs)中,Myr-Akt、H-RasG12V或K-RasG12D的表达增加了依赖mros的软琼脂集落形成[69]。此外,解除对Myc表达的控制也被证明可以改变ROS水平。Myc的外源性表达增加了ROS的生成,导致一些细胞发生转化,而ROS诱导其他细胞凋亡[70,71]。这表明活性氧效应可能依赖于细胞类型、其它突变和癌基因的表达水平。有趣的是,在小鼠癌症模型中,K-RasG12D、B-RafV619E或Myc生理表达的激活抑制了ROS[31]的稳态水平。这种抑制被证明是通过NRF2抗氧化计划的诱导介导的,因此,尚不清楚在这种情况下,癌基因是否通过增加抗氧化蛋白的表达来修饰ROS的产生,或者仅仅是降低稳态ROS。另一种可能是NRF2的表达抑制了细胞内ROS的总水平,但区域化的ROS(如mROS)的增加得以维持,从而促进了致瘤信号的传递。
靶向ROS治疗
抑制活性氧以抑制增殖
活性氧有助于有丝分裂信号,因此降低细胞内活性氧水平是抑制癌症生长的一种有吸引力的方法。有鉴于此,一些大规模研究调查是否与抗氧化维生素补充,包括β-carotene和维生素A和维生素E可以减少患癌症的风险。与预期结果相反,补充维生素d增加了两种情况下的癌症风险[96,97]。与这些结果一致的是,在K-Ras-或b
-
rafs诱导的肺癌遗传小鼠模型中,NAC或维生素E治疗可显著促进肿瘤生长和加速死亡[98]。这些结果表明,抗氧化剂在癌症治疗中的潜在应用是复杂的,在应用之前需要仔细验证。这些抗氧化剂作为癌症治疗失败的一种可能性是它们缺乏特异性。一般抗氧化剂对患者的治疗可能调节许多与癌症生长相关的生理过程。例如,作为癌症生长重要调节因子的免疫系统已被证明对ROS水平敏感[99]。另一种可能性是,普通抗氧化剂与靶向抗氧化剂的效果不同。线粒体靶向的抗氧化剂在体内外都被证明是有效的癌细胞生长抑制剂[69,100]。因此,需要进一步的研究来确定靶向抗氧化剂是否是治疗癌症的可行方法。
另一种抑制ROS的方法是减少ROS的产生。降低mROS的产生必然涉及抑制ETC,因此可能不实际,因为抑制线粒体呼吸固有的毒性。然而,服用降糖药二甲双胍的患者最近被证明有降低癌症发病率和死亡率的风险[101]。二甲双胍已被证明是ETC复合物I的抑制剂[102,103]。我们最近使用了一种二甲双胍不敏感复合物I模拟物来证实二甲双胍的抗癌作用主要是通过对体内癌细胞复合物I的特异性抑制来实现的[104]。有趣的是,我们还观察到,用二甲双胍治疗抑制缺氧HIF1α激活,表明它可能会减少生产的mro在缺氧。这种作用对二甲双胍的抑癌作用是否重要还有待进一步研究。另一种减少ROS生成的方法是抑制NADPH氧化酶。事实上,NADPH氧化酶4的缺失已经被证明可以激活胰腺癌细胞的凋亡[105]。此外,NADPH氧化酶活性抑制剂已被证明对体内癌症小鼠模型有疗效[106,107]。
增加活性氧选择性杀伤癌细胞
考虑到癌细胞已增加活性氧水平,它们可能对进一步增加活性氧的破坏作用具有选择性敏感性。增加癌症细胞中活性氧的具体产生可能是困难的,尽管这是目前多少化疗药物发挥作用的一种被提出的机制[108]。另外,由于癌细胞经常增加抗氧化剂的表达以维持体内平衡,一种有希望的治疗方法是抑制抗氧化剂,使癌细胞暴露于内生性产生的ROS[109]。为了支持这一模型,一些小分子筛选识别出了能够特异性抑制转化细胞生长的化合物,这些化合物集中在谷胱甘肽的利用上[110-112]。在所有病例中,经鉴定的小分子治疗降低了谷胱甘肽水平,增加了ROS,并且可以通过NAC治疗挽救。此外,抑制抗氧化途径也被证明是有效的抑制癌症生长。NRF2基因敲除可抑制胰腺癌和肺癌小鼠模型的疾病进展[31,32]。小分子ATN-224对SOD1的抑制在体外可导致ros依赖性癌细胞死亡,在体内可降低晚期k
-
ras驱动肺癌的肿瘤负荷[113]。这些最近的例子进一步证明,无论是通过增加ROS的产生还是抑制抗氧化剂,增加ROS都是一种很有前途的靶向癌细胞的方法(图6)。
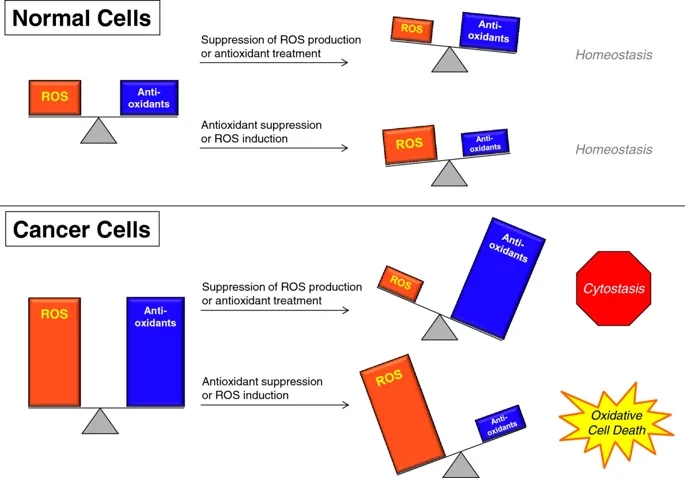
通过改变活性氧水平来靶向癌细胞。与癌细胞相比,正常细胞中活性氧和抗氧化剂的含量都减少了。因此,无论是活性氧还是抗氧化剂的丢失,都会导致活性氧稳态发生微小变化,使细胞存活并发挥功能。然而,由于癌细胞有更多的活性氧和抗氧化剂,它们可能更容易受到活性氧水平变化的影响。使用抗氧化剂或防止ROS生成会导致细胞失去足够的ROS信号来维持生长。其结果是细胞停滞和可能的衰老。另外,抑制抗氧化剂或增加活性氧的生成会导致癌细胞中活性氧过多,导致癌症特异性氧化细胞死亡。
结论
ROS在肿瘤发生生物学中的重要作用日益明显。虽然在这里已经提出了几种机制,但大量的ROS介导的信号靶点在很大程度上是未知的。然而,癌症相关突变增加ROS水平的频率表明,增加ROS的产生可能是癌基因和肿瘤抑制因子中大量癌症相关突变的共同结果。此外,线粒体突变的明显选择增加了活性氧,损害了代谢的灵活性,这表明活性氧在这些癌细胞中被强烈选择。一个新出现的模型是,癌细胞增加活性氧的产生来激活局部的致瘤信号,但是通过提高抗氧化活性来平衡活性氧的增加来维持氧化还原平衡。与所有关于癌症的研究一样,最终的目标将是设计能够利用这些发现的治疗方法。在这方面,通过抑制活性氧来阻止致瘤信号通路的激活和通过禁用抗氧化剂来诱导细胞死亡来加剧活性氧都是有希望的方法。今后的研究需要更好地了解以ROS为目标的途径。此外,未来的研究需要确定什么来源的活性氧和什么特定的抗氧化剂需要的稳态。有了这些知识,我们可以更好地理解癌症生物学,并设计出新的疗法来专门治疗癌细胞。
Mitochondrial reactive oxygen species and cancer
Abstract
Mitochondria produce reactive oxygen species (mROS) as a natural by-product of
electron transport chain activity. While initial studies focused on the damaging
effects of reactive oxygen species, a recent paradigm shift has shown that mROS
can act as signaling molecules to activate pro-growth responses. Cancer cells
have long been observed to have increased production of ROS relative to normal
cells, although the implications of this increase were not always clear. This is
especially interesting considering cancer cells often also induce expression of
antioxidant proteins. Here, we discuss how cancer-associated mutations and
microenvironments can increase production of mROS, which can lead to activation
of tumorigenic signaling and metabolic reprogramming. This tumorigenic signaling
also increases expression of antioxidant proteins to balance the high production
of ROS to maintain redox homeostasis. We also discuss how cancer-specific
modifications to ROS and antioxidants may be targeted for therapy.
Mitochondrial reactive oxygen species and cancer | Cancer & Metabolism | Full
Text
https://cancerandmetabolism.biomedcentral.com/articles/10.1186/2049-3002-2-17
肿瘤微环境中活性氧的种类:综述
Reactive Oxygen Species in the Tumor Microenvironment: An
Overview
摘要活性氧(ROS)是肿瘤重要的信号分子。ROS水平将决定生理效应。虽然高水平的活性氧会导致组织损伤和细胞死亡,但低水平的活性氧会产生增殖效应。ROS是由肿瘤细胞产生,也由构成肿瘤微环境(TME)的细胞成分产生。在这篇综述中,我们讨论了ROS影响TME的机制,重点是肿瘤浸润性白细胞。在这种情况下,对活性氧生物学的更深入的了解可能允许对活性氧水平进行治疗性操作,以重塑肿瘤微环境并增加抗肿瘤活性。
关键词:ROS;线粒体;肿瘤发生;肿瘤微环境;基质;组织浸润淋巴细胞;新陈代谢
1. 介绍
活性氧(ROS)包括超氧化物、过氧化氢和羟基自由基。虽然活性氧对脂质、蛋白质和DNA具有损伤作用,但近年来其作为重要的胞内和胞外信号分子的作用日益明显。线粒体是细胞内ROS的主要来源,在调节增殖、凋亡和代谢途径中起重要作用[2,3,4]。研究证实,肿瘤的特征包括代谢重编程和肿瘤促进微环境[5]。这两个重要的生物学事件的交界面都是由癌细胞以及微环境中的细胞成分产生的ROS[6,7,8,9]。理解细胞外和细胞内ROS之间的相互作用,不仅是在肿瘤内,而且与构成肿瘤微环境的细胞有关,对我们理解肿瘤发生的过程至关重要。在这篇综述中,我们将讨论活性氧在肿瘤发生中的作用。我们还将重点介绍ROS如何调节TME内细胞的生物学过程,包括癌症相关成纤维细胞(CAFs)和肿瘤浸润免疫细胞(TII)。
2. 活性氧在肿瘤发生中的作用
细胞内产生的内源性活性氧主要来源于线粒体或过氧体内的代谢反应。然而,也有一部分活性氧是由烟酰胺腺嘌呤二核苷酸磷酸(NAPDH)氧化酶(NOX)产生的,而NOX是跨膜蛋白家族的一种,它在生物膜上运输电子并催化氧转化为超氧化物。然后,超氧化物被超氧化物歧化酶(SODs)进一步还原生成H2O2。环氧合酶、脂氧合酶和胸苷磷酸化酶[10]也能产生活性氧。
ROS在肿瘤发生中发挥重要作用,影响细胞增殖、基因组不稳定性、炎症、抗凋亡和代谢重编程等多种生物学过程。活性氧水平的增加可以在许多癌细胞株中观察到。在肿瘤细胞中,活性氧主要由线粒体产生。线粒体通过线粒体电子传递链(ETC)[12],从氧的单电子还原中产生超氧化物(O2•)。在线粒体内,ROS在许多不同的位点产生,其中最重要的是复合物I、II和III[13,14]。复合物I和II在线粒体基质中产生O2•,复合物III在基质和膜间隙中都产生O2•[15,16]。线粒体基质生成的O2通过超氧化物歧化酶蛋白2
(SOD2)[17]转化为H2O2。复合物iii产生的膜间间隙O2•可以到达线粒体外膜并进入胞质,在胞质中被超氧化物歧化酶蛋白1
(SOD1)[18]转化为H2O2。由于能够接触到胞质,复杂的iii生成的ROS被认为是影响细胞信号转导[19]的原因。
如前所述,癌症中活性氧水平通常升高,然而,高水平的活性氧会产生有害的影响,因此,细胞已经进化出维持活性氧平衡的机制。这些机制包括过氧化清除系统(过氧化物酶),通过将H2O2还原为H2O[20]来控制H2O2水平。线粒体ROS的产生也受O2的可用性、通过ETC的电子通量率、给定电子载体的浓度和线粒体膜电位的调节[12,21]。最后,线粒体在细胞内的定位对影响细胞信号通路有重要影响,因为线粒体聚集到细胞的离散区域可以优先影响相邻的信号通路[22,23]。这些机制共同平衡了ROS的作用,肿瘤细胞可以利用ROS使细胞优先进入增殖状态。
线粒体ROS可以刺激多种信号通路。也许最广为人知的是,在缺氧条件下,线粒体ROS需要稳定缺氧诱导的转录因子(HIFs)[24,25,26,27]。HIF的稳定导致广泛的转录程序的启动,包括对血管生成[28]重要基因的调控。为了使血管生成发生,内皮细胞的增殖需要[29]。为此,HIF上调血管内皮生长因子(VEGF)的表达。VEGF是一种可溶性生长因子,与VEGF受体结合,激活对内皮细胞增殖至关重要的信号通路。VEGF的有丝分裂作用最常见的是通过激活细胞外信号调节的激酶/丝裂原激活的蛋白激酶(ERK/MAPK)途径介导的,该途径是一种强有力的细胞增殖促进因子[31]。线粒体ROS在激活t细胞[32]方面也很关键。小鼠复杂iii生成的线粒体ROS水平降低,导致在CD3或CD28[32]刺激下仍不能持续激活t细胞。此外,一项研究表明,线粒体在t细胞系中移位至免疫突触,线粒体H2O2需要通过MAPK信号[33]进行t细胞受体(T-cell
receptor,
TCR)信号转导。总之,这些数据表明,线粒体ROS在t细胞刺激和增殖所需的抗原刺激后增强TCR信号转导。总之,线粒体ROS在刺激生理细胞增殖方面发挥重要作用,可被肿瘤利用以促进生存和生长。
肿瘤产生高水平的活性氧[11]。最初,人们认为高水平的活性氧通过对DNA的氧化损伤导致基因组不稳定性[34]而导致肿瘤的发生。然而,研究也表明,癌细胞[35]中细胞抗氧化剂的蛋白表达增加。因此,癌细胞有能力在不引起大量氧化损伤的情况下维持高水平的有丝分裂信号。事实上,癌基因和/或肿瘤抑制因子在癌细胞中的丢失会导致ROS的产生。例如,在一项研究中,致癌的H-RasG12V在3T3成纤维细胞中过表达,表明有丝分裂信号[36]所需的ROS的产生增加。此外,p53抑癌基因Akt、H-RasG12V或KrasG12D的表达以及缺失p53对小鼠胚胎成纤维细胞的转化表明,软琼脂[6]中需要线粒体ROS才能实现无锚点生长。在许多人类癌症[37]中,线粒体DNA在几个基因中发生突变,这些基因对细胞的功能至关重要。这些突变也会导致线粒体ROS生成水平的增加[38,39,40]。在K-ras驱动的肺癌小鼠模型中,线粒体转录因子A
(TFAM)的缺失显示了肿瘤生长速度的降低。TFAM是线粒体DNA复制所必需的。当TFAM缺失时,氧化磷酸化受损,因此线粒体ROS水平降低。此外,这项研究表明,线粒体ROS所需锚固独立增长在许多癌症细胞类型[6],由肿瘤细胞ROS的产生中扮演一个重要的角色在推动肿瘤发生然而,ROS生产由其他肿瘤浸润细胞以及整个当地时间的氧化状态具有深远影响肿瘤生物学(图1)。
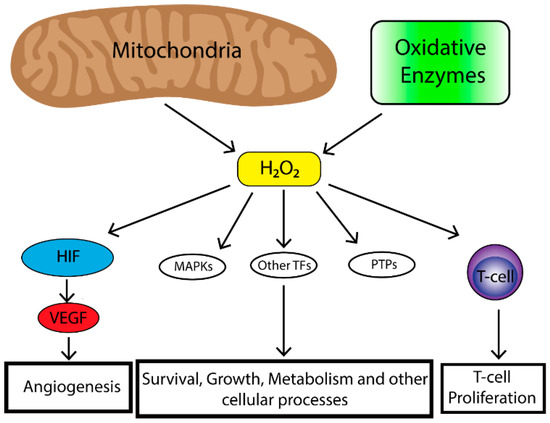
图1所示。线粒体是细胞活性氧(ROS)水平的主要贡献者,而氧化酶(如NAPDH氧化酶、环氧合酶、脂氧合酶和胸腺嘧啶磷酸化酶)也有助于细胞ROS的积聚。线粒体活性氧对细胞生物学有许多影响,包括增殖蛋白激酶(MAPK)(例如,extracellular-signal-regulated激酶(ERK),
p38
MAPK,小君n端激酶(物),诱导转录因子(例如,核因子kappa-light-chain-enhancer激活B细胞(NF-κβ),低氧诱导转录因子(HIF),激活蛋白1
(AP-1),核呼吸因子(NRF),热休克因子1
(HSF-1))和放松管制的蛋白质磷酸酶(例如,磷酸酶和紧张素同系物(PTEN)。这导致在HIF的情况下,通过MAPKs、转录因子和蛋白磷酸酶以及免疫细胞功能和调节,增强血管生成、生存、生长、代谢改变等细胞过程。
3.肿瘤相关成纤维细胞、活性氧和肿瘤微环境(TME)
TME不仅包括肿瘤细胞,还包括肿瘤淋巴管、肿瘤血管、细胞外基质、非肿瘤基质细胞以及化学调节剂(即,趋化因子,细胞因子,生长因子)和微生物种群。细胞外基质(ECM)和基质包括间质基质和基底膜,可作为许多刺激肿瘤发生的生长因子和趋化因子的储存场所。非癌性间质细胞包括内皮细胞、周细胞、免疫细胞、活化脂肪细胞、间充质干细胞、正常成纤维细胞和CAFs。正常的成纤维细胞负责细胞外基质的周转和组织内稳态。它们是伤口愈合和衰老过程的基础。与正常的成纤维细胞不同,CAFs可出现在肿瘤边缘或浸润到肿瘤内。与癌细胞相关的活化成纤维细胞被称为CAFs,在癌症的发生、发展和转移过程中起着关键作用[41,42]。CAFs进一步细分为成纤维细胞和肌成纤维细胞。Alpha-smooth肌肉肌动蛋白(α-SMA)阳性myofibroblasts是肿瘤中的主要的CAFs[43]。
.jpg)
CAFs的主要作用是增强肿瘤生成[44]。浸润的CAFs比正常的成纤维细胞更具增殖能力,并激活对促进肿瘤生长重要的特异性信号通路[45,46,47]。在体内[48]中,位于肿瘤边缘但不在肿瘤内的CAFs具有促进肿瘤进展的能力。已知这些CAFs可分泌CXCL12等因子,进而激活邻近上皮细胞[49]中的促肿瘤通路,如AKT。几乎所有的实体肿瘤都存在CAFs。在某些肿瘤如乳腺、胰腺和前列腺中,CAFs可占肿瘤质量的80%,因为它们导致了纤维组织或结缔组织(desmoplasia)[50]的过度生长。癌组织中高比例的CAFs与预后不良、肿瘤相关巨噬细胞和上皮向间质转化(EMT)[50]的浸润增加有关。
纤维组织增生是肿瘤进展的标志,并产生机械力,通过压迫血管来限制淋巴和血液供应,从而形成缺氧环境[51]。此外,这些机械力可以导致成纤维细胞转化成肌成纤维细胞[51]。如前所述,缺氧刺激线粒体ROS的产生,癌细胞产生的ROS水平高于正常组织,影响CAF功能[9,52]。CAFs可以来源于上皮细胞、内皮细胞、造血干细胞、周细胞或脂肪细胞,以及基质组织中的常驻成纤维细胞[53,54,55,56,57,58,59]。大部分腺癌的CAFs表达平滑肌α-actin(α-SMA),因此,被称为myofibroblasts [60]。肌成纤维细胞的主要功能是伤口愈合和修复,在肿瘤中,这些细胞可以作为紊乱的慢性伤口愈合的驱动因素。几项研究表明,ROS可以作为肌成纤维细胞分化的驱动因素。几项研究报道了ROS在成纤维细胞向肌成纤维细胞转变中的重要性(图2A)。转化生长因子β1 (TGF-β1)和基质细胞衍生因子1 (SDF-1)和其他人发挥重要作用在推动myofibroblast从纤维母细胞的转变。众所周知,mitochondrial-ROS所需TGF-β1激活。实际上,当纤维母细胞受到mitochondrial-ROS药理抑制剂,TGF-β1表达水平降低[61]。SDF-1也可以诱导成肌成纤维细胞向肌成纤维细胞的转化,这种转化依赖于ros[58,60]。此外,暴露于慢性氧化应激的成纤维细胞也可分化为肌成纤维细胞[9,60]。从氧化应激小鼠模型中分离出的关键抗氧化转录因子被耗尽的成纤维细胞显示出向肌成纤维细胞的转化,而长期使用外源性抗氧化剂可以逆转这种转化[60,62]。此外,由于前列腺癌成纤维细胞中谷胱甘肽过氧化物酶3和硫氧还蛋白还原酶I等抗氧化酶的上调,导致ROS水平下降,从而抑制其向肌成纤维细胞的分化[63]。综上所述,ROS可促进人肿瘤中肌成纤维细胞的分化。
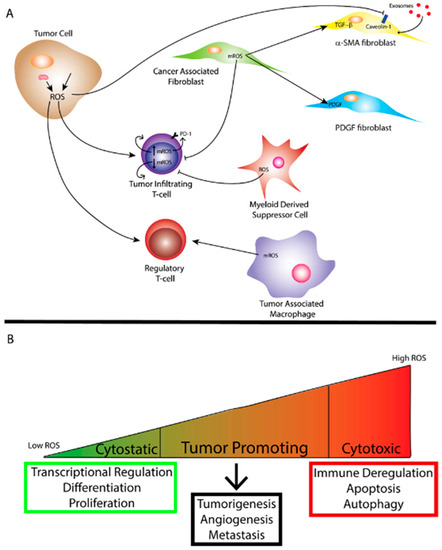
图2。(A)肿瘤细胞内线粒体和/或外源性来源产生的活性氧(ROS)影响肿瘤免疫,从而促进更具致瘤性的环境。线粒体ROS
(mROS)可刺激癌相关成纤维细胞(CAFs)的分化,肿瘤细胞产生的ROS可通过抑制caveolin-1促进外泌体的摄取,从而导致某些CAFs的代谢重编程。ROS也可根据mROS水平影响肿瘤浸润t细胞的功能。骨髓来源的抑制细胞(MDSCs)和肿瘤相关巨噬细胞(TAMs)也会产生ROS,影响其他免疫细胞的功能,ROS也会影响t细胞的调节功能。(B)活性氧的数量对应于对生物功能的不同影响。ROS的细胞抑制水平导致生物过程的维持,而细胞毒性水平的ROS导致细胞死亡和免疫解除。当活性氧达到超生理或细胞抑制水平时,肿瘤就会通过活性氧促进生长,同时避免细胞死亡。如前所述,氧化应激可以从肿瘤细胞中产生。
ROS还可影响CAFs的增殖和迁移。如前所述,CAFs是一个异质性群体,不同的群体有不同的标记表达。虽然α-SMA
myofibroblasts代表大多数的战乱国家还有其他标记,用于检测成纤维细胞[9]的亚型。然而,这些亚型是否真正代表不同的成纤维细胞亚群尚不清楚。研究表明,ROS可以影响成纤维细胞亚型[9]。成纤维细胞的一个亚型,可能受到活性氧的影响血小板源生长因子β(PDGF-β)成纤维细胞。ROS通过抑制磷酸酶在PDGF信号转导中发挥重要作用[64,65,66,67]。PDGF信号的激活刺激成纤维细胞的生长和运动。有趣的是,一项研究表明,当PDGF刺激正常的人成纤维细胞时,NOX4和DUOX4这两种负责增加细胞内ROS水平的酶会调节细胞周期进入[68]。这些研究表明,在PDGF-β成纤维细胞ROS可能发挥不可或缺的作用影响成纤维细胞增殖和迁移。另一种可能受活性氧影响的成纤维细胞标记物是Caveolin-1。研究表明,当成纤维细胞和肿瘤上皮细胞在氧化应激下共培养时,成纤维细胞中ca
-1的降解可以通过抗氧化剂和自噬抑制剂的治疗来阻止[69,70,71]。综上所述,这些研究表明成纤维细胞产生的ROS在CAF的激活和分化中发挥重要作用。然而,肿瘤细胞也会大量产生ROS等代谢副产物,并在CAF功能中发挥作用。如前所述,CAFs产生的ROS也可以增强肿瘤的发生。
H2O2由肿瘤上皮细胞产生,可扩散至其他组织和细胞。H2O2也参与了细胞内信号通路的激活。某些研究侧重于了解H2O2如何影响肿瘤微环境(TME)和间质。在这些研究中,乳腺癌细胞与CAFs共培养,以证明肿瘤产生的H2O2对CAFs的影响。有趣的是,肿瘤H2O2导致线粒体功能降低,葡萄糖摄取增加,活性氧增加[70,72]。此外,共培养的癌细胞线粒体活性增加,GLUT1表达降低,葡萄糖摄取降低[70]。肿瘤细胞与CAFs之间的这种交互作用可以通过添加过氧化氢酶来消除[70]。最后,与乳腺癌细胞共培养的成纤维细胞显示出腔蛋白-1
(Caveolin-1)的下调和肌成纤维细胞标志物的表达增加[70]。这表明,肿瘤细胞产生的活性氧可以直接重组CAFs,潜在地创造一个更具有致瘤性的微环境。
小凹蛋白,如cave -1,是在多种细胞类型中发现的独特的蛋白质,有助于形成小凹,这是一种质膜内陷。ca
-1的表达是通过自我消化或自噬介导的[73,74]。与正常成纤维细胞相比,人类CAFs的CAV-1表达通常较低。CAV-1表达减少与糖酵解增加和线粒体功能降低有关,这种CAV-1表达的减少被认为是由肿瘤细胞氧化应激诱导的自噬介导的[8,74,75]。抗氧化剂和自噬抑制剂可以消除成纤维细胞介导的CAV1降解[70,71]。CAV-1表达不仅参与了代谢开关的诱导,还参与了自噬/mitophagy活性和微环境[9]的重构。有趣的是,不同类型活性氧对肺癌细胞中ca
-1的表达有不同的影响。例如,羟基自由基上调了ca -1的表达,而O2•和H2O2下调了ca -1的表达。还应注意的是,ca
-1的降解导致细胞外泌体摄取增加[76]。
外泌体是细胞外小泡的一个亚型,由腔内内小泡衍生而来。外泌体由脂质双分子层组成,包含蛋白质、mrna、脂质、mirna和自由代谢物,内化后释放到靶细胞胞质中[77]。肿瘤来源的EVs能够在肿瘤微环境中转化非恶性细胞,从而促进肿瘤的发生[78,79]。因此,EVs为细胞串扰提供了一种机制。确实,研究表明外泌体具有对受体细胞重新编程的能力,能够调节受体细胞的增殖、存活和免疫效应状态[80]。最近,Zhao等人证明,从前列腺癌和胰腺癌患者来源的CAFs中分离出的外泌体可以抑制癌细胞线粒体氧化磷酸化,增加糖酵解和还原性羧化[81]。
外泌体通过不同的途径进入细胞,外泌体进入细胞的过程存在争议。最近的一项研究表明,来自胶质母细胞瘤(GBM)细胞的外泌体通过非经典的脂筏依赖性内吞作用内化[76]。作者随后证明,脂筏相关蛋白,ca
-1,负调控外泌体的摄取[76]。此前有报道称,肿瘤细胞可诱导氧化应激,导致细胞自噬降解ca
-1[8,74]。总之,这些研究表明了肿瘤细胞产生ROS的途径,ROS向成纤维细胞发出信号,导致ca
-1降解,从而增加外泌体摄取。外泌体摄取的增加会导致代谢物的流入增加,并导致成纤维细胞的代谢重新编程,形成更具致瘤性的CAF。(图2A)。事实上,Zhao等人证明,从胰腺癌和前列腺癌CAFs中分离出的外泌体含有大量的谷氨酰胺、乳酸和乙酸盐,以及许多其他氨基酸和代谢物,表明外泌体在无胸膜症和脂肪生成中起作用[81]。
自噬是将细胞质细胞器或组分隔离到自噬小体中,并将其传递到溶酶体进行降解的途径。自噬对生存、分化、发育和代谢至关重要,并参与许多疾病状态,如癌症。细胞应激包括活性氧可以刺激自噬[82,83]。ROS能够直接和间接调节自噬[84,85,86]。ros诱导的自噬已被证明可以保护细胞免受氧化损伤,这表明ros依赖的负反馈环可以调节细胞内的氧化应激[87]。有缺陷的自噬存在于多个肿瘤中,支持肿瘤抑制作用[88,89,90]。然而,研究也表明自噬具有促进肿瘤的功能,这意味着自噬功能在癌症中是依赖于环境的[91,92]。除了癌症类型,这种依赖于环境的功能可能也适用于TME中的细胞。目前有证据表明,ROS可以通过自噬在CAFs和肿瘤细胞之间提供交互作用,从而创造一个更具致瘤性的环境。一项研究在乳腺癌的异种移植模型表明,HIF-1α-dependent激活自噬的基质细胞致瘤性提高[93]。鉴于HIF-1α线粒体活性氧的作用,它可以猜测,在基质细胞ROS可能通过诱导自噬调节肿瘤细胞的致瘤性。另一项研究探索了从卵巢癌组织中分离出的CAFs以及从良性组织中分离出的正常成纤维细胞,发现CAFs具有抗氧化应激的能力,并且这一过程是通过自噬介导的[94]。CAFs可以作为TME内氧化应激的中心介质,帮助肿瘤细胞规避TME
ROS水平升高的细胞毒性效应。肿瘤细胞也可以通过自噬和mitophagy影响TME细胞,即通过自噬选择性降解线粒体。几项研究表明,肿瘤细胞可诱导CAFs内的代谢增加,而CAFs内的代谢增加又诱导自噬和mitophagy,使重要的生物分子和代谢前体得以循环利用[95,96]。预期其副产品也会在CAFs中生成ROS,这也有助于将其重新编程为更具致瘤性的成纤维细胞。
如前所述,TME涉及大量不同类型的细胞,包括肿瘤浸润性白细胞(TILs)。TILs包括骨髓来源的抑制细胞(MDSCs)、肿瘤相关巨噬细胞(TAMs)和调节性T细胞(Tregs)以及其他免疫细胞。MDSCs、TAMs和treg可同时抑制肿瘤的免疫反应。研究表明,免疫抑制的微环境允许更大的肿瘤侵袭、转移和对治疗的耐药性[97]。虽然已知ROS是肿瘤进展的促进剂,但它们可能通过免疫细胞抑制实现这一点(图2A)。在本节中,我们将主要关注ROS对肿瘤浸润T细胞的影响。T细胞对于宿主对癌症的免疫反应至关重要。肿瘤浸润的细胞毒性T细胞。CD8+ T细胞)在抗肿瘤免疫反应中起关键作用。然而,在癌症进展过程中,肿瘤微环境变得免疫抑制和T细胞毒性被抑制。如前所述,ROS也与更具免疫抑制作用的肿瘤微环境相关。ROS在微环境中与T细胞的激活和调节有关,扮演着多种角色。高水平的ROS在TME中抑制T细胞增殖和抗肿瘤功能。此外,肿瘤微环境中其他细胞产生的ROS导致癌症患者的T细胞低反应性[98]。另外,T细胞激活、增殖和功能需要较低水平的ROS[99,100]。T细胞的活化是通过刺激TCR和共刺激受体来实现的,共刺激受体可诱导信号通路和转录因子的产生。此外,依赖于tcr的钙离子流入CD4+ t细胞,导致CD4+ t细胞激活所需的线粒体ROS生成[32,101]。ETC的复合物I和III都被认为是t细胞激活所必需的线粒体ROS的来源[32,102]。肿瘤浸润T细胞显示线粒体功能的损失可以通过表达PGC1α获救,线粒体生物起源的关键球员,随后恢复抗肿瘤活性[103]。此外,最近的一项研究表明,在透明细胞肾细胞癌肿瘤中存在CD8 TILs,但它们在功能和代谢方面受到损害[104]。它们被发现产生大量的线粒体ROS,导致线粒体超氧化物歧化酶2 (SOD2)的下调[104]。线粒体ROS清除剂MitoQ和MitoTEMPO可以挽救这种效应,CD8 TIL活化的增加证明了这一点[104]。另一项研究表明,经bicionic表达载体嵌合Ag受体(CAR)共表达过氧化氢酶(CAT)修饰的T细胞,细胞内氧化应激降低,CAR-CAT T细胞以抗原特异性方式裂解肿瘤细胞的能力增强[105]。有趣的是,与传统的CAR- cat t细胞不同,这些CAR- cat t细胞能够在细胞外氧化应激下发挥作用[105]。CAR T细胞依赖于肿瘤特异性TCRs和CAR的遗传转移,从外周血转移到T细胞。不幸的是,在实体肿瘤中,CAR t细胞不仅要达到它们的目标,而且还必须在不利的肿瘤微环境中生存和发挥作用。上述研究强调了理解活性氧生物学以影响生物学结果的重要性。
PD-1是一种主要由激活的T细胞表达的表面受体,是免疫反应的负调控因子。PD-1与两种配体(PD-L1或PD-L2)中的一种结合,导致其受体的磷酸化和Src同源区2域内含磷酸化酶-2
(SHP-2)的募集,从而导致TCR激活分子的去磷酸化[106,107]。这导致了肿瘤的“暴露”和肿瘤反应性细胞毒性t淋巴细胞(t -
ctl)的激活。PD-1阻断如何导致肿瘤耐受的消除仍是一个很大的未知数。然而,最近发表的一篇论文表明,ROS和线粒体激活在PD-1阻断诱导T细胞依赖性抗肿瘤活性中发挥作用[108]。在本研究中,作者发现,从PD-L1阻断处理的小鼠中分离出的T-CTLs具有线粒体质量、膜电位、超氧化物和细胞ROS的增加,表明这些细胞具有更高的线粒体代谢率[108]。此外,作者发现,使用ROS发生器(叔丁基过氧化氢)或线粒体呼吸链解耦物(FCCP)处理这些细胞,可以进一步协同PD-1阻滞对肿瘤生长抑制的作用[108]。这表明,调节T-CTLs等免疫细胞中的线粒体活性和线粒体ROS可能对PD-1阻断反应产生深远影响。可能PD-1抑制剂无应答者的线粒体激活水平较低,因此ROS水平较低,从而导致t
- ctl的次优激活。
另一项研究表明,小鼠肿瘤细胞系对PD-1阻断的敏感性取决于细胞的耗氧和缺氧能力[109]。作者发现,二甲双胍处理细胞时,体内外耗氧量被抑制,从而导致瘤内缺氧[109]。二甲双胍联合PD-1阻断可改善小鼠瘤内t细胞功能并清除肿瘤[109]。这表明,较低水平的ROS,从而较低的低氧肿瘤环境,可以提高PD-1阻断免疫治疗的疗效。因此,可以想象,肿瘤微环境中整体ROS和缺氧水平的降低,加上特定肿瘤浸润细胞中细胞内线粒体ROS的增加,可能会对PD-1阻断产生最有效的反应。
所述研究的二分法强调了理解线粒体ROS如何调节到不同水平以影响生物学结果的重要性(图2A)。活性氧的阴和阳可能是免疫肿瘤学领域的治疗靶点,因为不同水平的活性氧对完全不同的免疫程序有抑制作用(图2B)。最近,针对PD-1/PD-L1阻断的免疫治疗改变了癌症治疗的格局,显著提高了癌症患者的生存率。目前,该疗法已被批准用于治疗黑色素瘤、非小细胞肺癌(NSCLC)、肾癌、霍奇金淋巴瘤和头颈癌。然而,值得注意的是,虽然在许多使用PD-1/PD-L1阻断治疗的患者中持久反应显著,但约有30-50%的患者对PD-1/PD-L1阻断仍无反应或反应较慢[110
- 111,112]。了解如何提高对PD-1/PD-L1阻滞的反应对患者的生存有显著的影响。
调节性t细胞(treg)也存在于TME中,并提供另一种免疫抑制来源,降低肿瘤反应性细胞毒性t细胞免疫。研究表明,免疫抑制的treg和细胞毒性t细胞之间的平衡可能受到代谢调节[113,114,115,116]。如前所述,肿瘤微环境中的ROS和氧化应激通过对肿瘤浸润免疫细胞的作用来促进肿瘤免疫。最近的一篇论文表明,肿瘤环境中ROS和氧化应激的增加导致Tregs更强的免疫抑制作用[117]。作者发现,凋亡的treg在体内外抑制t细胞活化的效率更高[117]。此外,这些凋亡的treg蛋白在荷瘤小鼠模型中破坏了PD-L1的治疗效果[117]。这被认为是通过释放大量具有免疫抑制作用的ATP和腺苷来介导的[117]。这增加了在肿瘤微环境中靶向ROS以降低ROS水平进而抑制Tregs免疫抑制活性的治疗可能性。在这一点上,最近的一项研究表明,线粒体ROS水平的降低导致Treg分化的减少[118]。Kunisada等人证明二甲双胍通过叉头盒P3
(Foxp3)蛋白抑制幼稚CD4+
t细胞向treg分化,从而减少肿瘤浸润treg的数量[118]。此外,作者还证明二甲双胍可诱导treg的代谢重编程,使其进入糖酵解状态[118]。二甲双胍是一种复合物I抑制剂,因此减少了复合物I生成线粒体ROS的池。二甲双胍处理的T-regs中线粒体ROS的减少可能也对改变代谢重编程重要的转录程序产生影响。此外,Weinberg等人最近的一项研究发现,Treg抑制活性需要线粒体等复合体III[119]。通过小鼠实验,作者证实Rieske铁硫蛋白(RISP)的缺失导致了Treg抑制功能的缺失,而Rieske铁硫蛋白是线粒体复合体III的一个重要亚基。复合物III产生的ROS是重要的信号转导物质[3,120]。特定的线粒体靶向抗氧化剂存在[6,121],观察这些药物是否反映了RISP敲除Tregs的效果将是很有趣的。基于上述研究,可能通过二甲双胍或线粒体靶向抗氧化剂降低线粒体ROS水平,treg的免疫抑制作用减弱,使细胞毒性t细胞的杀瘤作用增强。
综上所述,这再次表明,特定细胞类型内的ROS水平对该细胞的功能有重要影响。如前所述,CTL中ROS水平高可能具有抗肿瘤作用,而TReg中ROS水平低可能与免疫抑制降低有关。此外,在不同的细胞类型中,类似水平的活性氧也可能产生相互矛盾的作用。如上图所示,在肾细胞癌的CD8
TILs中,高水平的ROS导致损伤和缺乏抗肿瘤反应,而在结肠癌小鼠的TILs中,高水平的ROS与增强的肿瘤杀伤作用相关。需要对肿瘤内ROS如何浸润免疫细胞以及细胞外ROS如何参与调节肿瘤免疫进行更深入的研究,以进一步确定不同水平、位置和类型的ROS如何影响肿瘤免疫。
也有证据表明,其他TIL如MDSC和TAMs是由ROS调节的。MDSCs是肿瘤微环境中主要的免疫抑制细胞类型之一[122]。这些细胞通过抑制T细胞来诱导免疫抑制。最近的一项研究表明,肿瘤诱导的MDSCs可以抑制T细胞增殖,通过产生ROS促进结直肠癌细胞生长[123]。在另一项研究中,使用ROS抑制剂可以完全消除MDSC对t细胞的免疫抑制作用[124]。值得注意的是,ROS通过抑制t细胞受体(T-cell
receptor,
TCR)与MHC-peptide复合物的识别来抑制t细胞免疫反应。这一点在一项研究中得到了强调,该研究表明,在ROS抑制剂过氧化氢酶(catalase)存在的情况下,MDSCs与t细胞共同培养,会破坏MDSCs介导的t细胞增殖[125]。最后,重要的是要了解,虽然高水平的ROS具有免疫抑制作用,但低水平的ROS可能对t细胞的激活很重要[126]。TAMs是肿瘤微环境中存在的另一类免疫细胞。TAMs被认为是炎症和肿瘤发生的关键介质。活性氧参与巨噬细胞的活化和信号传导。此外,有研究表明巨噬细胞来源的ROS可诱导treg[127]。这提示巨噬细胞来源的活性氧具有免疫抑制作用。另一项研究表明,ROS需要促进更多侵入性表型tam与黑色素瘤,这种效应是通过ROS-dependent介导肿瘤坏死因子α分泌[128]。值得注意的是,本研究的作者发现黑素瘤TAMs在多个线粒体生物发生和呼吸链基因中表达升高,表明线粒体ROS是TAMs内氧化应激的主要来源之一[128]。这些研究强调了ROS不仅是氧化应激的诱导因子,而且是肿瘤微环境中免疫调节的介质,在促进肿瘤发生方面具有重要作用。
5. 微生物组和活性氧
构成TME的另一个元素是微生物组。在过去的几年里,许多研究都围绕着宿主微生物群之间的关系,以及微生物群落内环境平衡的破坏(生态失调)是如何影响诸如癌症等病理条件的。众所周知,宿主微生物群可通过诱导促炎毒素、改变信号通路或破坏抗肿瘤免疫功能等途径促进致癌[129,130,131,132]。宿主微生物群可能诱发致瘤状态的另一种方式是通过产生ROS[133]。例如,粪肠球菌是一种共生菌株,它可以产生大量的胞外超氧化物,转化为H2O2,从而破坏真核细胞的DNA[134]。致病性脆弱拟杆菌产生毒素,上调细菌多胺分解代谢途径,产生可导致DNA损伤和结肠肿瘤形成的活性氧[135]。进一步的研究表明,某些种类的细菌利用胆汁酸进行呼吸,产生可导致胃肠道癌症的dna损伤ROS副产品[136,137]。另外,损伤的粘膜依赖氧化还原信号和活性氧来修复。微生物群产生和排出甲酰化肽,这些肽激活结肠上皮甲酰肽受体,诱导局部ROS生成,激活对上皮伤口愈合重要的信号通路[138]。综上所述,这说明了ROS在微环境中的两分法作用:既是损伤因子,又是生长和愈合促进剂。为了了解ROS是如何完全影响TME的,还需要进行更多的研究来探索细菌种特异性ROS在肿瘤、免疫和基质细胞功能中的具体作用。
虽然许多数据支持TME中微生物区系衍生的ROS在肿瘤发生中的作用,但研究发现宿主线粒体代谢的变化与宿主微生物区系的变化相关[139]。Yardeni等人最近的一项研究表明,宿主线粒体通过ROS影响肠道微生物群落多样性[140]。通过检查小鼠肠道菌群中改变线粒体功能的基因的各种突变,作者能够表明,线粒体遗传变异改变了肠道菌群的组成。进一步分析与肠道菌群改变相关的线粒体DNA变异表明,多样性与宿主ROS的产生有关。此外,他们能够证明,小鼠体内ROS水平的调节导致肠道菌群的改变。作者发现,线粒体ROS的减少导致肠道菌群中物种多样性的增加。最近的一项研究表明,对免疫治疗有反应的黑色素瘤患者肠道菌群多样性增加[141]。综上所述,这表明线粒体ROS的调节可用于增强癌症患者对免疫治疗的敏感性。
6. ROS靶向的治疗意义
如前所述,活性氧处于一个十字路口,可能连接肿瘤和免疫微环境。因此,ROS作为一种调控肿瘤和微环境的交互作用和改善肿瘤预后的手段,是一种很有吸引力的治疗靶点。最近,针对线粒体复合物I(线粒体ROS生成的重要物质)的研究已经获得了临床应用。10多年前,二甲双胍被广泛应用于2型糖尿病的治疗中,发现其可降低糖尿病患者的癌症风险[142]。进一步的研究,探索二甲双胍的作用机制,证实了二甲双胍在体外抑制复合物I的能力[143,144,145]。进一步研究表明,在异种移植小鼠模型中,二甲双胍靶向线粒体复合物I可抑制复合物I,减少复合物I生成ROS并减少肿瘤发生[146]。这导致了大量的临床研究,迄今为止产生了令人失望的结果。目前,有多个临床试验正在进行或积极招募围绕二甲双胍治疗癌症。这些新试验主要涉及二甲双胍联合免疫治疗(NCT03311308、NCT03874000、NCT03994744)、化疗(NCT02122185、NCT01310231、NCT03238495、NCT02122185、NCT03243851)、强力霉素(NCT02874430)和间歇性禁食(NCT03709147)在特定的癌症患者亚群中的应用。也有重要证据表明二甲双胍可能通过调节TME来影响肿瘤的进展[147]。通过调节复合物I
ROS和线粒体代谢,二甲双胍具有改变TME细胞群表型的能力。进一步的研究有可能解决二甲双胍与特定的细胞因子抑制剂(如IL-6、IL-17或FOXP3)结合PD-1/PD-L1更有效地重建TME的问题。值得注意的是,二甲双胍是一种弱复合物I抑制剂,目前正在使用更强、更特异的线粒体ROS抑制剂进行其他临床研究。在第一阶段的一项研究中,我――344年,一个isoflavone-derived复杂我线粒体抑制剂,正在评估早期her2阴性乳腺癌患者结合贝伐单抗,一个anti-vascular血管内皮生长因子(VEGF-A)抑制剂,评估我-
344和贝伐单抗是否能抵消所发生的代谢变化与vegf治疗(NCT02806817)。抗血管生成治疗的这些代谢变化与耐药性有关。众所周知,化疗诱导ROS生成,随着时间的推移,肿瘤对化疗产生耐药性。在二线治疗中或在一线治疗的特定时间点使用二甲双胍可能潜在地消除或延长耐药性的发展。最后,其他临床研究的重点是寻找不仅阻断complex
I,也阻断complex III等的靶向途径,而complex
III被认为是线粒体ROS的主要产生者,对细胞信号通路的诱导非常重要。一项这样的研究正在慢性骨髓性白血病(CML)患者中进行,使用的抗生素是Tigecycline,它会破坏线粒体DNA翻译,随后抑制ETC复合物I、III、IV和V的形成(NCT02883036)。在未来,了解和重新利用对线粒体ROS生成有特殊影响的药物可能会导致联合治疗,从而改善癌症患者的预后。
治疗干预的另一个潜在靶点是TCA周期。癌症有代谢紊乱,现在很清楚线粒体代谢是肿瘤生长所必需的。近年来的研究主要集中在针对特定的TCA循环酶。其中一种药物是CPI-613,它是一种脂酸盐类似物。硫辛酸是丙酮酸脱氢酶和-酮戊二酸-脱氢酶复合物的辅因子,两者都是TCA循环中限速步骤所必需的。这些酶的抑制导致线粒体失调。正如预期的那样,这种损伤导致线粒体ROS的诱导[148]。CPI-613诱导线粒体ROS有可能使肿瘤细胞内氧化还原平衡向细胞毒性方向倾斜(图2B)。CPI-613也可能对TME中的免疫细胞产生影响。例如,免疫抑制的免疫细胞如M2巨噬细胞、Tregs和MDSCs依赖于氧化磷酸化和线粒体代谢。CPI-613可能以这些促肿瘤细胞类型为靶点,使氧化还原平衡向细胞功能障碍或毒性方向倾斜。已经有转移性胰腺癌患者的激动人心的I期数据表明,CPI-613联合标准护理化疗显著提高了整体反应率和潜在的无进展生存[149]。目前,有几项临床试验探索CPI-613联合化疗用于多种不同的肿瘤亚型(NCT02168140、NCT02232152、NCT03699319、NCT02484391)。在未来,将CPI-613与免疫治疗相结合可能是一个潜在的研究领域,以及了解如何将CPI-613用于产生耐药性的患者。
最后,另一个有吸引力的治疗靶点是抗氧化机制。最近,RTA-408,也被称为omaveloxolone,被研究用于黑色素瘤。RTA-408是一种半合成的三萜,已知可诱导核因子、红细胞2相关因子2
(Nrf2),后者是保护机体免受氧化或亲电应激的主要细胞调节因子[150,151,152]。RTA-408通过这一机制抑制已在肿瘤异种移植物中得到证实的活性氧和氮[153]。RTA-408的这种作用也在MDSCs中被证明[153]。在较高浓度下,RTA-408还通过核因子kappa-B激酶亚基选择性地抑制肿瘤生长[153]。该药物可同时靶向肿瘤和免疫抑制的TME。目前,RTA-408联合免疫疗法治疗黑色素瘤患者的临床I/II期研究正在进行中。
总之,靶向肿瘤内ROS和TME有可能改善目前的治疗结果。虽然单独靶向ROS不太可能获得治疗效果,单药二甲双胍试验证明了这一点,但与细胞毒性或免疫调节剂联合使用有可能显著改善反应和生存。了解如何调节敏感的氧化还原平衡内和之间的肿瘤和不同的tme浸润免疫细胞可能导致更有效的治疗。未来,细胞特异性活性氧调制器可能成为优化以活性氧为靶点的潜在治疗方法的关键。
7. 结论
ROS在维持健康细胞内的生理稳态中起着重要作用。在肿瘤细胞中,会产生促生长、促致肿瘤环境的活性氧生成途径被利用。目前已知TME不仅包括肿瘤细胞,还包括非肿瘤间质细胞如CAFs以及肿瘤浸润性免疫细胞,在肿瘤与环境的交互作用中发挥重要作用,从而驱动肿瘤的发生。越来越清楚的是,代谢重编程和随后的ROS生成是这种串扰的关键。不同水平的活性氧对肿瘤细胞及其微环境内生物结果的差异影响,对我们理解肿瘤的发生、生长和进展越来越重要。了解如何利用活性氧来调节细胞特异性的生物学功能,不仅有助于开发新的抗癌治疗方法,而且可能提高现有治疗方法的疗效。
血管病理学中的NADPH氧化酶
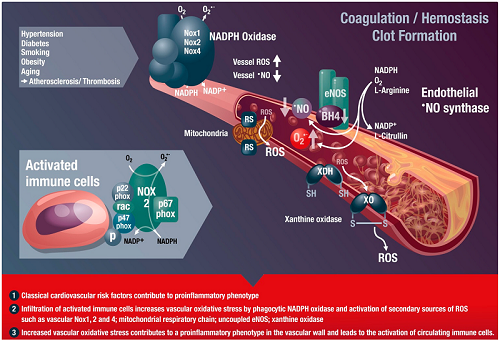
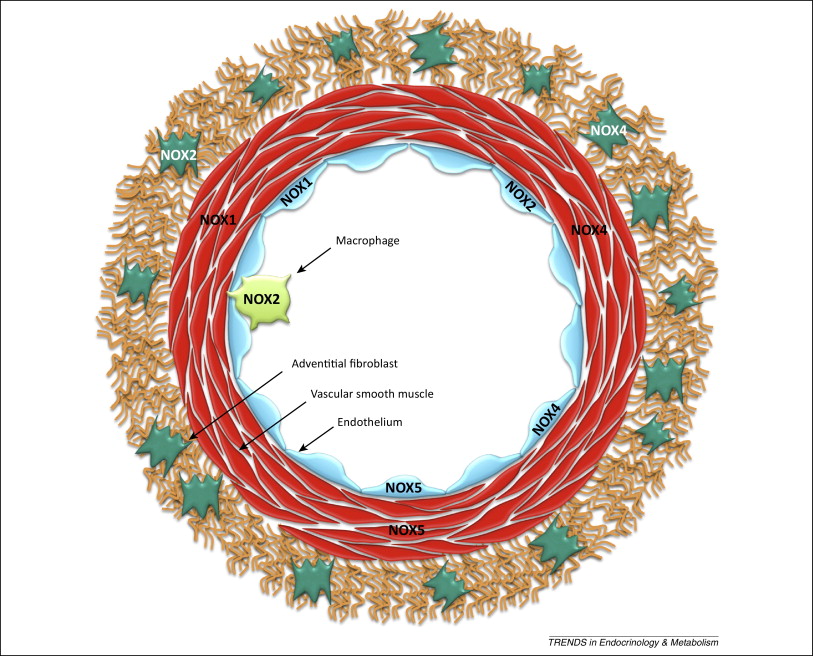
意义:活性氧在血管疾病中起重要作用。虽然活性氧有许多可能的来源,但烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶起着核心作用。它们是“点燃自由基”的来源,而自由基会影响其他酶,如一氧化氮合酶、内皮型一氧化氮合酶或黄嘌呤氧化酶。这很重要,因为动脉粥样硬化(高血压、糖尿病、高胆固醇血症和吸烟)的危险因素调节血管壁NADPH氧化酶的表达和活性。最新进展:哺乳动物中有七种亚型:Nox1、Nox2、Nox3、Nox4、Nox5、Duox1和Duox2。Nox1、Nox2、Nox4和Nox5在内皮细胞、血管平滑肌细胞、成纤维细胞或血管周围脂肪细胞中表达。其他同系物未被发现或表达水平很低;他们的作用还没有确定。Nox1/Nox2可促进内皮功能障碍、高血压和炎症的发展。Nox4可能在应激时起到保护血管系统的作用;然而,当它的活性增加时,它可能是有害的。钙依赖的Nox5参与了人动脉粥样硬化的氧化损伤。关键问题:NADPH氧化酶衍生的ROS在血管病理和维持正常生理血管功能中发挥作用。我们还讨论了最近阐明的机制,如NADPH氧化酶在血管保护、血管炎症、肺动脉高压、肿瘤血管生成、中枢神经系统调节血管功能和高血压中的作用。未来方向:了解单个氧化酶和同源物在血管疾病中的相互作用,对于在实验室和临床有效地调节血管NADPH氧化酶是至关重要的。Antioxid。氧化还原信号,20,2794
- 2814。
NADPH Oxidases in Vascular Pathology
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4026218/