凋亡信号通路与淋巴细胞稳态 Apoptosis signaling pathways and lymphocyte homeostasis
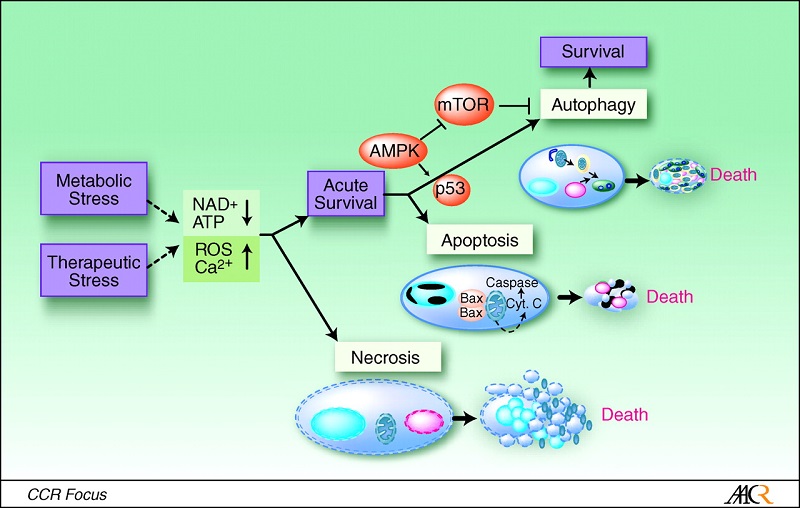
The relationship between necrosis, autophagy, and apoptosis. Metabolic and therapeutic stresses lead to acute NAD+ and ATP depletion accompanied by increased intracellular calcium and ROS. Cells that do not adapt to these changes undergo necrotic cell death. The activation of stress regulators, such as AMPK, allows cells to acutely survive these changes. AMPK-dependent phosphorylation results in the inhibition of mammalian target of rapamycin, which inhibits autophagy. AMPK-dependent phosphorylation also activates p53, which can lead to autophagy or apoptosis, through the activation of Bax and Bak, the cytoplasmic release of cytochrome c (Cyt. C), and the activation of caspases. Unlike apoptosis or necrosis, stress-induced autophagy can lead to autophagic cell death or to cell survival.
http://clincancerres.aacrjournals.org/content/13/24/7271
The relationship between necrosis, autophagy, and apoptosis. Metabolic and therapeutic stresses lead to acute NAD+ and ATP depletion accompanied by increased intracellular calcium and ROS. Cells that do not adapt to these changes undergo necrotic cell death. The activation of stress regulators, such as AMPK, allows cells to acutely survive these changes. AMPK-dependent phosphorylation results in the inhibition of mammalian target of rapamycin, which inhibits autophagy. AMPK-dependent phosphorylation also activates p53, which can lead to autophagy or apoptosis, through the activation of Bax and Bak, the cytoplasmic release of cytochrome c (Cyt. C), and the activation of caspases. Unlike apoptosis or necrosis, stress-induced autophagy can lead to autophagic cell death or to cell survival.
Oxidative post-translational modification of cysteine residues in proteins. Cysteine is commonly located on the surface and at the active site of proteins, either alone (monothiols) or in close proximity to another cysteine residue (vicinal dithiols). Vicinal dithiols tend to form disulfides upon oxidation, whereas monothiols undergo reversible oxidation to sulfenic acid. Under strongly oxidizing conditions, sulfenic acid is further oxidized to sulfinic and sulfonic acids. Other modifications that also occur include acetylation, glutathionylation, nitrosylation, and carbonylation of protein cysteines. These changes can result in alterations in protein-protein interactions, enzyme activity, DNA and/or RNA binding, and membrane interactions. HNE, 4hydroxynonenal; ROS, reactive oxygen species.
Oxidative post-translational modification of cysteine residues in... | Download Scientific Diagram
https://www.researchgate.net/figure/Oxidative-post-translational-modification-of-cysteine-residues-in-proteins-Cysteine-is_fig1_270654660
Bcl-2调控的凋亡途径
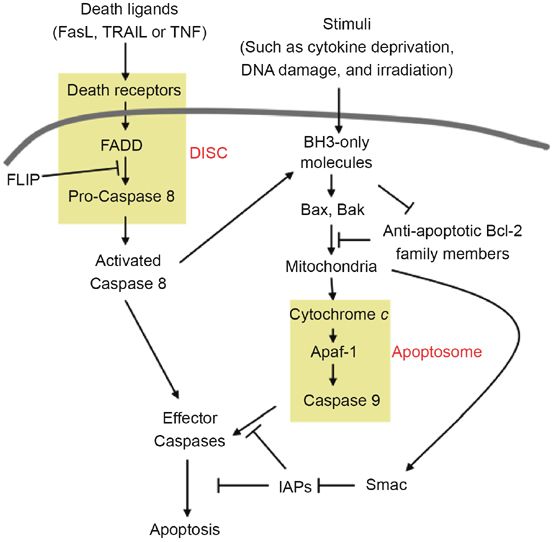
F4: Programmed cell death type I: Apoptosis: In response to cellular damage, activation of p53 results in increased expression of Bax and antagonism of the anti-apoptotic protein Bcl-2. As a result, mitochondrial membrane permeability increases and cytochrome C is released into the cytosol. Cytochrome C binds Apaf-1 to form an apoptosome complex leading to activation of caspase-9 and initiation of the caspase cascade and resultant cell death.
Programmed cell death type I: Apoptosis: In response to | Open-i
https://openi.nlm.nih.gov/detailedresult?img=PMC3055228_1476-4598-10-12-4&req=4
Apoptosis and Necrosis
Mark P. Mattson, ... Scott T. Brady, in Basic Neurochemistry (Eighth Edition), 2012
Necrosis
Necrosis is a dramatic and very rapid form of cell death in which essentially every compartment of the cell disintegrates
Necrosis is characterized by marked dysregulation of ion homeostasis resulting in cell swelling, dilation of mitochondria and the ER and the formation of vacuoles in the cytoplasm. Proteases play important roles in the degradation of cells during necrosis. In contrast to apoptosis, where caspases are the key death proteases, calpains and lysosomal proteases (cathepsins B and D, in particular) are major players in necrosis. Caspases may be activated in response to mitochondrial damage and cytochrome c release during necrosis but appear not to be essential for cell death. During the cell death process the chromatin clumps and the nuclear membrane is disrupted. Finally, the cell lyses, releasing its contents into the extracellular compartment, where the contents may damage neighboring cells and induce an inflammatory response. Transcription of genes and protein synthesis stop and ATP is rapidly depleted in cells undergoing necrosis.
There are few cell death triggers that are only capable of inducing either apoptosis or necrosis
Instead, whether a cell undergoes apoptosis or necrosis is usually determined by the intensity and/or duration of the death-inducing stimulus. In general, severe and/or sustained insults trigger necrosis whereas less severe transient stresses induce apoptosis. For example, moderate over-activation of glutamate receptors may trigger apoptosis while more intense and sustained activation of glutamate receptors induces excitotoxic necrosis (Chs. 17 and 35). The environment of the cell at the time it is subjected to the death stimulus can also determine the mode of death. Thus, a certain level of glutamate receptor activation may induce apoptosis in a cell receiving normal amounts of oxygen and glucose but may cause necrosis when that same cell is subjected to ischemia.
Trauma
Acute trauma as in head injury (e.g., automobile accidents, sports injuries) can induce necrosis of the tissue at and surrounding the site of the trauma. Physical damage to cellular membranes may induce necrosis in the traumatized tissue, while disruption of ion homeostasis and energy depletion may trigger necrosis in cells adjacent to the directly damaged cells. Traumatized cells release the contents of their organelles into the extracellular space, resulting in exposure of adjacent cells to various lysosomal proteases and to acidosis. Marked changes in pH can also trigger necrosis; interestingly, intracellular acidification may contribute to necrotic cell death induced by extensive DNA damage.
Energy failure/ischemia
Cellular energy failure is one defining feature of necrosis, and severe reduction in glucose and/or oxygen availability is sufficient to trigger necrosis. Mitochondrial toxins can trigger apoptosis at low levels but at higher levels they induce necrosis as the result of severe depletion of ATP.
Excitotoxicity
Overactivation of glutamate receptors, particularly when neurons are subjected to metabolic and oxidative stress, can trigger excitotoxic necrosis. Sodium ion influx through AMPA/kainate receptors and voltage-dependent sodium channels induces cell swelling, and calcium ion influx through NMDA receptors and voltage-dependent calcium channels activates various proteases (calpains, for example) that degrade various structural and metabolic proteins.
Calcium ion release from the ER and the accumulation of misfolded proteins in the ER can trigger necrosis, and agents that inhibit such calcium release can protect cells against necrosis.Necrosis - an overview | ScienceDirect Topics
https://www.sciencedirect.com/topics/neuroscience/necrosis
Expression of Bcl-2 increases intracellular glutathione by ...
https://www.ncbi.nlm.nih.gov/pubmed/9703946
Jul 30, 1998 · Overexpression of Bcl-2 and related anti-apoptotic gene products has been shown to increase the intracellular concentration of the antioxidant tripeptide glutathione in neuronal and hematopoietic cells.
Cited by: 125
Publish Year: 1998
Author: Michael J. Meredith, Carrie L. Cusick, Syrus Soltaninassab, Konjeti S. Sekhar, Shelly Lu, Michael L....
[PDF]Inhibition of Glutathione Synthesis Reverses Bcl-2 ...
https://cancerres.aacrjournals.org/content/canres/63/2/312.full.pdf
suggest that Bcl-2-mediated cisplatin resistance in MCF-7 cells is depend- ent on up-regulation of glutathione production, which contributes to cell survival by mechanisms independent of cisplatin inactivation or inhibition of DNA adduct formation.
Cited by: 224
Publish Year: 2003
Author: Charles M. Rudin, Zejia Yang, Lisa M. Schumaker, DavidGlutathione in Cancer Cell Death - PubMed Central (PMC)
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3756414
Mar 11, 2011 · Bcl-2-AS therapy has been evaluated in multiple clinical trials [172-174]. Bcl-2-AS therapy using G3139, for example, an 18-base phosphorothioate oligonucleotide complementary to the first six codons of the Bcl-2 mRNA, selectively and specifically inhibits Bcl-2 expression and promotes apoptosis in different human and murine cancer cell lines .
Cited by: 191
Publish Year: 2011
Hydrogen Peroxide-Induced Akt Phosphorylation Regulates ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2697849
Reactive oxygen species such as hydrogen peroxide (H 2 O 2) are involved in many cellular processes that positively and negatively regulate cell fate.H 2 O 2, acting as an intracellular messenger, activates phosphatidylinositol-3 kinase (PI3K) and its downstream target Akt, and promotes cell survival.The aim of the current study was to understand the mechanism by which PI3K/Akt signaling ...
Cited by: 75
Publish Year: 2009
Author: Mahdieh Sadidi, Stephen I. Lentz, Eva L. Feldman
Catalpol inhibits apoptosis in hydrogen peroxide-induced ...
https://www.researchgate.net/publication/40449956_Catalpol_inhibits_apoptosis_in...
Catalpol inhibits apoptosis in hydrogen peroxide-induced endothelium by activating the PI3K/Akt signaling pathway and modulating expression of Bcl-2 and Bax ... reduced intracellular ROS release ...
The selection between apoptosis and necrosis is ...
www.nature.com/articles/4401249
Jul 17, 2003 · A factor that may trigger cell death, and that may decide the selection between apoptosis and necrosis, is the increase in intracellular oxidation. Using treatment with exogenous hydrogen peroxide (H 2 O 2) as an experimental paradigm of oxidation, it was often observed that moderate H 2 O 2 concentrations triggered apoptosis,...
Published in:
Cell Death & Differentiation · 2003
Authors:
Alfonso Troyano · Patricia Sancho · Carlos Iglesias Fernandez · E De Blas · Paolo Berna…
Affiliation:
Spanish National Research Council · University of Padua
About:
Necrosis · Cisplatin · Apoptosis
https://www.nature.com/articles/nchembio0905-188
凋亡(程序性细胞死亡)是去除感染的,受损的或有害的细胞所必需的,其破坏的调节与癌症,自身免疫和变性疾病有关。在分子水平上,多个信号传导途径汇聚在一个半胱氨酸蛋白酶(胱天蛋白酶)家族上,该半胱氨酸蛋白酶被激活时会通过裂解一系列重要的细胞底物而引起细胞破坏。 Bcl-2蛋白家族是导致许多(但不是全部)信号导致caspase活化的关键调节因子。在这里,我们着重介绍了它们的功能,许多著名的评论对此进行了广泛的讨论(参见Borner,2003; Cory和Adams,2002; Cory等,2003; Gross等,1999; Strasser等,2000)。
从蠕虫到人的细胞死亡机制的进化保护
Bcl-2相关蛋白构成了秀丽隐杆线虫和哺乳动物等多种物种中保守的核心凋亡机制的一部分。在功能上,Bcl-2相关蛋白抑制或促进细胞凋亡,而属于相对派系的蛋白之间的相互作用决定了细胞是存活还是死亡。也许最容易理解的途径是蠕虫线虫,详细的遗传研究表明,两种Bcl-2相关蛋白(促凋亡EGL-1和促存活CED-9)对于控制发育程序化的体细胞是必不可少的死亡(Horvitz,1999)。死亡信号EGL-1的表达是由损伤信号诱导的。 EGL-1与蠕虫Bcl-2直系同源基因CED-9的结合从CED-9释放衔接子蛋白CED-4。一旦释放,CED-4就会结合并激活caspase CED-3,从而导致细胞死亡。
保守的领域是家庭的特征
哺乳动物具有至少五个存活前CED-9的同源物,即Bcl-2,Bcl-xL,Bcl-w,Mcl-1和A1:它们均在发育过程中以及对细胞应激的反应中抑制细胞凋亡。它们与CED-9共享至少三个保守的BH(Bcl-2同源性)结构域。这些中的三个(BH1,BH2和BH3)折叠在一起,以在前生存分子上形成疏水凹槽(Muchmore等,1996)。该沟是促凋亡EGL-1或其哺乳动物对应物Bik / Nbk / Blk,Bid,Bad,Hrk / DP5,Bim / Bod,Noxa,Puma / Bbc3,Bmf和Bcl-Gs结合配体的靶标。这些杀手蛋白(仅BH3蛋白)具有较短的BH3结构域,但没有其他明显的序列,具有更宽的Bcl-2家族(Huang和Strasser,2000)。仅BH3蛋白质与其同源伴侣的结合是通过两亲性α-螺旋BH3结构域形成的疏水面与存活蛋白的疏水沟(由BH1-BH3形成)之间的相互作用而发生的(Petros et al。,2000 ; Sattler等,1997)。除了仅BH3蛋白外,另一类促凋亡Bcl-2蛋白是多结构域Bax样蛋白。它们与它们的亲生存表亲具有显着的序列和结构相似性(Suzuki et al。,2000),但反而起到促进细胞死亡的作用,这可能与仅BH3蛋白不同。仅BH3蛋白质:细胞福祉的传感器
生化和遗传实验表明,仅BH3的蛋白质可作为细胞死亡的触发因子(Huang和Strasser,2000)。例如,EGL-1或Bim的丢失会导致发育过程中的细胞过多,并且它们对许多损伤信号具有抵抗力。为了防止不适当的细胞死亡,仅通过多种机制来控制仅BH3的蛋白质,直到细胞受到的伤害解除制动为止(Puthalakath和Strasser,2002)。在秀丽隐杆线虫的雌雄同体特定神经元中,TRA-1A抑制EGL-1的转录,直到其被发育线索除去(Conradt and Horvitz,1999)。对于Hrk / DP5,Bim,Noxa和Puma也已描述了仅对哺乳动物BH3蛋白的转录控制。后两种蛋白可能是肿瘤抑制蛋白p53响应DNA损伤而诱导的细胞死亡的关键介体(例如Nakano和Vousden,2001)。也已经描述了通过螯合或蛋白质修饰的转录后对照。 Bim和Bmf被隔离到细胞骨架结构中,应激信号可能通过诱导其磷酸化来触发其释放(Lei和Davis,2003年)。相反,存活因子通过促进其与14-3-3支架蛋白的结合的磷酸化作用使健康细胞中的Bad失活。全长出价似乎没有什么活性,但是被半胱天冬酶裂解产生活性的C端片段(tBid),当通过N末端的肉豆蔻酰化靶向线粒体时,它是有效的杀手。
半胱氨酸蛋白酶激活,细胞死亡执行者
不同的仅哺乳动物BH3的蛋白质与独特的(但重叠的)应激信号传导途径偶联。当破坏信号去除施加在其上的正常对照时,它们被释放以结合并灭活存活前的Bcl-2蛋白。由于仅BH3蛋白靶向位于核被膜,内质网和线粒体外膜的胞质面上的前生存性Bcl-2蛋白,因此这些蛋白的位置可能部分地调节了这种细胞转换。 Bcl-2蛋白在线粒体上的作用最受关注,因为当线粒体外膜被破坏时,线粒体膜间空间会释放促细胞凋亡因子,例如细胞色素c,Smac / Diablo和HtrA2 / Omi(Martinou)和格林(2001)。细胞色素c触发与迄今发现的唯一的哺乳动物CED-4同源物Apaf-1和启动子胱天蛋白酶caspase-9一起激活下游胱天蛋白酶的全酶('凋亡小体')的形成(Zou et al。,1997)。 。然而,哺乳动物存活前的Bcl-2蛋白不具有直接隔离Apaf-1的功能,与秀丽隐杆线虫CED-9螯合CED-4不同(Moriishi等,1999)。此外,在许多细胞类型中,由应激信号激活的细胞色素c / Apaf-1 / Caspase-9途径似乎并不是caspase活化的唯一途径(Marsden等,2002)。因此,Bcl-2必须控制线粒体细胞色素c释放之前(上游)的步骤(Cory和Adams,2002)。
多域Bax样蛋白
多结构域Bax样亚家族可能负责破坏线粒体外膜,允许释放诸如细胞色素c的因子,尽管这种发生方式仍存在争议(Kuwana等,2002; Roucou等,2002)。 。 Bax和Bak在功能上是多余的,因为单独丢失任何一种蛋白质都会导致轻度异常,但它们的综合丢失会导致多余组织的明显积累(Lindsten等,2000)。有趣的是,也废除了仅BH3蛋白的杀伤作用(Cheng等,2001; Zong等,2001),但尚不清楚仅BH3蛋白是否直接作用于Bax / Bak来激活它们。这样的模型似乎很有吸引力,因为Bax还带有疏水沟,可能是仅BH3蛋白结合并因此被杀死的真正目标。 BH3结合可能促进胞质Bax的膜移位,其构象改变和聚集,与Bax激活相关的事件。令人惊讶的是,很少有数据支持仅BH3蛋白直接结合Bax / Bak(Wang等,1996; Wei等,2000)或它们在含Bax / Bak的复合物中的存在(Antonsson等,2001; Nechushtan等)。等人,2001)。另一种可能性是,存活前的Bcl-2-样蛋白直接或间接控制Bax / Bak激活:BH3结合消除了该活性,从而允许Bax / Bak激活。
粘性尾巴在通往粘性终点的道路上
BH3结合如何消除存活前Bcl-2的活性?除了由BH1-BH3在靶向BH3结合的存活前Bcl-2上形成的保守的疏水沟外,大多数存活前分子还具有C端疏水区。尽管此区域可能充当跨膜结构域,允许膜插入,但生存前Bcl-w的3D结构表明疏水尾巴通常不暴露,而是占据了仅BH3配体靶向的疏水沟(Denisov等, 2003; Hinds等人,2003)。 BH3结合将取代尾部,从而允许更紧密的膜相互作用,并且此步骤使存活前的分子失活(Wilson-Annan等,2003)。已经提出了一种类似的机制来激活Bax作为其C末端,这是生物活性所必需的,并且通常不会暴露出来(Suzuki等,2000)。如前所述,释放Bax尾巴及其膜易位的直接触发机制尚不清楚(Guo等,2003)。了解仅BH3蛋白如何使Bcl-2失活以及如何导致Bax / Bak激活可能是了解这些蛋白功能的关键。
观点
自从克隆Bcl-2并发现其生物学功能以来(Vaux et al。,1988),Bcl-2蛋白家族已被证明是细胞凋亡的关键调节剂,并且在了解基本分子方面已取得了巨大进展。细胞死亡控制的机制。但是,仍然存在许多问题。每一类蛋白质的功能是否相同(Nijhawan等,2003)?生存前Bcl-2如何发挥作用? Bcl-2及其存活前同源物(如秀丽隐杆线虫CED-9)是否直接控制哺乳动物CED-4直系同源物,因为蠕虫中人Bcl-2的过度表达可以补偿CED-9的丧失(Hengartner和霍维茨(1994)?或者,Bcl-2是否直接控制线粒体和其他细胞内膜的完整性(Kaufmann等,2003; Scorrano等,2003)?这个蛋白质家族能否通过影响膜融合和裂变而间接控制膜的完整性(Karbowski等,2002)? Bcl-2是否仅充当仅BH3蛋白质的“沉陷”,从而阻止Bax / Bak活化(仅BH3配体的真正靶标)(Letai等人,2002)?或者,Bcl-2能否通过其他机制控制Bax / Bak的激活?如果是,为什么人类Bcl-2在没有明显Bax样直系同源物的蠕虫中发挥功能?仅BH3蛋白是仅通过与生存前Bcl-2结合而杀死,还是尚未确定其靶标?随着更多古老的,必不可少的,最终引人入胜的细胞过程被揭露,未来几年将为进一步的洞察提供令人兴奋的可能性。
Apoptosis signaling pathways and lymphocyte homeostasis | Cell Research
https://www.nature.com/articles/cr200752
凋亡信号通路与淋巴细胞稳态
抽象
自“细胞凋亡”一词首次被提出来描述一种独特的细胞死亡形式以来,已经过去了近三十年,这种细胞死亡涉及有序的,依赖基因的细胞分解。现在已经公认,凋亡是后生动物必需的生命过程,并且对于组织和器官的形成和功能至关重要。在成年哺乳动物体内,凋亡对于免疫系统的正常运作特别重要。近年来,随着分子和细胞生物学的迅速发展,在理解导致细胞凋亡的机制方面已取得了很大的进步。一般认为,凋亡细胞死亡诱导有两种主要途径:通过死亡受体的外在信号传导导致死亡诱导信号复合物(DISC)的形成,以及通过线粒体的内在信号传导主要导致线粒体的形成。凋亡的。 DISC或凋亡小体的形成分别激活执行凋亡过程的引发剂和常见效应子胱天蛋白酶。在免疫系统中,两种途径都起作用。然而,尚不知道它们是否足以维持淋巴细胞稳态。最近,已显示出淋巴细胞中存在新的凋亡机制,包括不依赖胱天蛋白酶的途径和粒酶引发的途径。这篇综述将总结我们对控制各种淋巴细胞群体稳态的机制的理解。凋亡细胞死亡
凋亡是细胞死亡的一种独特形式1,表现出特定的形态和生化特征,包括细胞膜起泡,染色质凝集,基因组DNA片段化以及特定吞噬信号分子在细胞表面的暴露2。经历凋亡的细胞不同于因坏死而死亡的细胞。坏死细胞通常被免疫系统识别为危险信号,从而导致炎症。相反,凋亡死亡是安静而有序的。据信,后生生物中大多数细胞死亡是通过细胞凋亡发生的。细胞凋亡对于器官,四肢和其他身体结构的形成以及维持成年体内大多数系统的功能至关重要。因此,凋亡信号传导过程的失调通常会导致严重的后果,例如神经退行性疾病3,癌症4或自身免疫5。
我们对控制细胞凋亡的分子机制的初步了解来自使用线虫秀丽隐杆线虫6、7、8、9、10的早期研究。然后将这些机制扩展到哺乳动物系统。现在已经鉴定出许多与细胞凋亡的起始,扩增或抑制有关的蛋白质和酶。在大多数细胞中,凋亡触发因素通常会导致胱天蛋白酶的活化,从而最终介导细胞的自毁。胱天蛋白酶通常以无活性的前体(称为蛋白酶)的形式存在,它们被裂解产生活性形式。已知的胱天蛋白酶的优选切割位点是在四个氨基酸基序X-X-X-Asp(其中X可以是任何氨基酸)的C-末端。活化的胱天蛋白酶依次切割各种细胞内和细胞质膜底物11,导致细胞崩解。
导致哺乳动物系统凋亡的主要途径有两个:由死亡受体引发的外在途径和通过线粒体发生的内在途径(图1)。外源性途径取决于适当的外源性介质与细胞表面死亡受体的结合。相反,内在途径响应来自细胞内的信号,例如由辐射和各种化学治疗剂引起的损害,以通过释放线粒体因子来诱导凋亡信号传导。
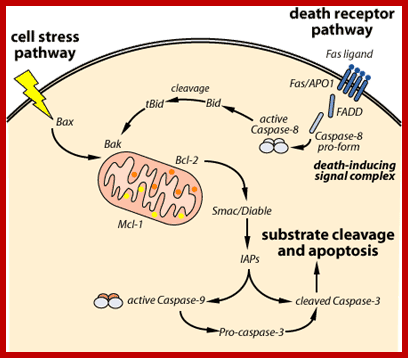
细胞凋亡途径的化学示意图。左侧显示了外在的凋亡信号通路,右侧显示了内在的凋亡信号通路。这些途径在胱天蛋白酶激活时收敛。 DISC和凋亡小体以黄色突出显示。
全尺寸图片
外源性凋亡信号通路
当细胞表面的死亡受体遇到特定的同源“死亡配体”时,就会启动细胞凋亡信号传导的外在途径,从而诱导通过细胞膜传递的构象变化。这些受体可以在配体结合的几秒钟内激活胱天蛋白酶,并在数小时内导致凋亡细胞死亡。已经描述了三种主要的特异性细胞死亡受体/配体对,即肿瘤坏死因子受体超家族(TNFRSF)的所有成员:(1)Fas和Fas配体(FasL)(Fas也称为Apo-1,CD95或TNFRSF6 12; FasL也称为CD178,CD95L或TNFSF6 13); (2)“死亡受体”(DR4和DR5)和TNF相关的凋亡诱导配体(TRAIL,也称为Apo2L或TNFSF10)14、15; (3)TNFα和TNF受体(TNF-R1)。这些死亡受体是TNFRSF的所有成员,是I型整合受体,具有一个保守的细胞外结构域,其中包含2至4个富含半胱氨酸的伪重复序列16,一个跨膜区域和一个保守的细胞内“死亡域”(DD),约为与衔接子蛋白相互作用的80个氨基酸17。
TNFR超家族的凋亡诱导受体以预先形成的三聚体形式存在于细胞表面。死亡受体的连接通过共同的转录/翻译非依赖性途径导致凋亡:配体结合诱导了死亡诱导信号复合物(DISC)的形成,而后者又裂解并激活了启动子胱天蛋白酶(胱天蛋白酶8或10)。引发剂胱天蛋白酶进而通过在特定位点的蛋白水解切割激活第二组胱天蛋白酶,称为效应子胱天蛋白酶。激活后,效应子胱天蛋白酶通过关键细胞内底物的降解达到凋亡过程。
Fas-FasL途径
在所有细胞死亡受体中,Fas的研究最为广泛。 Fas及其与膜结合的同源配体FasL的特异性相互作用触发了Fas三聚体的聚集。这使得Fas胞质尾巴中的DD能够立即募集DISC18。DISC由Fas,一种称为Fas相关死亡域(FADD)19的辅助蛋白和前胱天蛋白酶8组成。介导包含DDRS的TNFRSF 20成员的信号传导。FADD带有一个N末端死亡效应域(DED)和一个C末端DD。响应受体聚集,FADD由Fas募集,这种相互作用通过两种蛋白中高度保守的DD基序进行协调。 FADD和Fas通过其C末端DD的相互作用揭示了FADD的N末端DED,使其能够将前胱天蛋白酶8募集到Fas信号复合体21、22中。功能性FADD蛋白的存在对于Fas介导的至关重要凋亡作为FADD显性阴性形式的表达完全消除了Fas诱导的细胞死亡23。此外,体内研究显示,缺乏FADD 24的小鼠淋巴细胞死亡显着减弱。DISC的形成触发了proDD的自我切割-半胱天冬酶8转变为活性半胱天冬酶8。活化的半胱天冬酶8(也称为FADD样IL-1转换酶(FLICE))随后裂解下游的前半胱天冬酶3、6和7。活化的半胱天冬酶3裂解多种细胞底物,包括DNA修复酶,细胞和核结构蛋白以及核酸内切酶抑制剂。此外,半胱天冬酶3具有激活其他半胱天冬酶的能力,例如通常以其酶原形式存在的半胱天冬酶6和7,导致细胞破坏的放大,从而确保完全执行Fas诱导的细胞凋亡。
除了FADD和前胱天蛋白酶8以外的分子也可能在Fas介导的凋亡中募集。受体相互作用蛋白(RIP)25,具有DD(RAIDD)26的RIP相关ICH(ICE和ced-3同源物)/ CED-3-同源蛋白和半胱天冬酶原2 27形成了Fas-的另一个信号级联介导的死亡途径。 RIP-RAIDD路径可以用作FADD-caspase 8系统的备份。但是,它通常不代表Fas介导的主要死亡途径
Fas介导的凋亡的每一步都受到调控。 FasL基因在大多数细胞中均无转录活性。 FasL表达的调节控制FasL / Fas介导的生物学效应,例如CD4 + T细胞的活化诱导的细胞死亡(AICD)。 Fas表达的诱导仅需要TCR激活的PKC,而FasL表达则需要激活PKC和NFAT,后者通过Ca2 +动员28激活。Fas表达的调节还控制Fas反应,例如在p53诱导的细胞凋亡中。膜结合的FasL刺激Fas可以被人类29中的可溶性诱饵受体DcR3,缺乏跨膜和/或DD的各种Fas亚型和可溶性的无活性FasL拮抗。 Fas-DISC复合物的caspase 8激活能力主要受称为FLICE样抑制蛋白(FLIP)26、30、31、32、33的抑制蛋白调节。FLIP存在几种与caspase 8结构相似的同工型。 ,但缺乏酶促活性。30.将FLIP整合到死亡受体的DISC中会禁用DISC介导的加工,从而阻止caspase的激活830。此外,Fas介导的凋亡受线粒体细胞死亡调节剂的控制。例如,一些Bcl-2家族成员或凋亡蛋白(IAPs)抑制剂通过这种途径(见下文)。
Fas介导的T细胞死亡不仅通过凋亡发生,而且通过坏死发生。 Fas介导的坏死需要FADD和RIP,而caspase 8似乎是可有可无的34。但是,尚不清楚Fas,FADD和RIP在处理坏死中的分子机制。尽管Fas主要被认为是死亡诱导物,但它也触发T细胞35中的增殖信号。
TRAIL-DR途径
像其他TNF家族配体一样,TRAIL以II型膜结合蛋白的形式存在,或者可以通过半胱氨酸蛋白酶从膜上裂解产生可溶形式。人和小鼠都具有五个TRAIL受体:死亡诱导受体DR4 36和DR5 9、30、37、38、39和诱饵受体DcR1 30、36、39、40,DcR2 40、41、42和OPG (骨保护素)43.在这些受体中,只有DR4和DR5具有介导凋亡诱导的功能性DD。诱饵受体没有。 DcR1缺少DD和跨膜域。它通过糖磷脂酰肌醇尾锚定在膜上。 DcR2具有截断且不起作用的DD。 OPG以二聚体可溶形式分泌,也可与RANKL结合。这些诱饵受体与TRAIL结合,但不转导信号,从而通过中断功能性死亡受体三聚体的形成来保护细胞免受TRAIL诱导的细胞凋亡。 DR4和DR5下游的DISC形成和Bid切割类似于Fas诱导的细胞凋亡。简而言之,TRAIL触发DR4或DR5的聚类,这会聚集FADD和前胱天蛋白酶8形成DISC。在激活胱天蛋白酶8之后,效应子胱天蛋白酶随后被激活。
细胞结合的TRAIL和基因工程可溶形式均迅速诱导了多种来源多样的转化细胞系的凋亡14、41。由于正常细胞表达诱饵受体,而肿瘤细胞不表达,因此认为TRAIL可以特异性杀死肿瘤。细胞,但这仍然存在争议。有报道表明TRAIL治疗可能引起肝毒性44、45,并且发现重组TRAIL可以诱导肝细胞凋亡45。但是后来的研究发现重组TRAIL的肝毒性是由于外源序列标签46引起的。
与非哮喘患者相比,在哮喘患者的上皮,气道平滑肌,血管平滑肌和整个间质组织中,TRAIL表达显着增加。在哮喘患者中还观察到TRAIL诱饵受体DcR2的表达增加和TRAIL受体DR4和DR5的表达降低。有人提出,抗原攻击后调节TRAIL和TRAIL受体的相互作用可能对促进哮喘患者嗜酸性粒细胞的存活至关重要47。
TNFα-TNFR1途径
TNFα是多功能促炎细胞因子。它以三个157个氨基酸的肽链的同型三聚体形式存在。 TNFα通过两个受体发挥作用:TNFR1(也称为p55),其包含DD 48、49、50和TNFR2(也称为p75),其缺乏DD 51、52。TNFR1信号传导的初始步骤涉及TNF三聚体与TNF-R1胞外域的结合以及抑制蛋白(死亡域的沉默子)从TNF-R1的胞内域释放。产生的聚集的TNF-R1细胞内结构域被衔接蛋白TNF受体相关的死亡域(TRADD)识别,该结构域募集RIP,TNF-R相关因子2(TRAF2)和FADD。后面的这些蛋白质向TNF-R1募集关键酶,这些酶负责启动信号传导事件。 Caspase 8被FADD募集到TNF-R1复合物中,在其中被自身裂解激活,并引发蛋白酶级联反应导致凋亡。 TRAF2募集细胞凋亡蛋白1(cIAP-1)和cIAP-2的细胞抑制剂。还认为TRAF2可以在受体附近或附近的复合物中激活MAPKKK,例如细胞外信号调节激酶激酶激酶1(MEKK1)或ASK1,从而激活一系列激酶,从而激活JNK。蛋白激酶RIP对转录因子NF-κB的功能激活至关重要。
TNFR1诱导的细胞凋亡涉及两个顺序的信号复合物。首先,TNF与TNFR1的结合募集TRADD,RIP1和TRAF2,以形成质膜结合复合物(由TNFR1,TRADD,RIP1和TRAF2组成的复合物I),并迅速发出NF-κB的信号。形成复合物I后,TRADD和RIP1被修饰并与TNFR1分离,然后TRADD(和RIP1)与FADD结合,导致caspase 8/10募集,形成细胞质复合物(复合物II,由TRADD,RIP1,FADD和caspase 8/10)并导致凋亡。 FLIP级别确定后一种形式是否复杂。当复合物I成功激活NF-κB时,细胞FLIP水平会充分升高以抑制复合物II的形成并阻止细胞凋亡。如果复合物I的初始信号未能激活NF-κB,则复合物II发出凋亡信号53。
TNFα连接的生物学结果取决于NF-κB和JNK信号转导之间的平衡:NF-κB促进存活,而JNK增强细胞死亡。 TNFα介导的JNK激活加速了NF-κB诱导的抗凋亡蛋白c-FLIP的更新。 JNK介导E3泛素连接酶Itch的磷酸化和激活,后者特异性泛素化c-FLIP并诱导其蛋白酶体降解。 JNK1或Itch缺乏症或JNK抑制剂治疗可使小鼠在TNFα诱导的急性肝衰竭的三种不同模型中产生抗药性,并且这些小鼠的细胞未显示出可诱导的c-FLIP泛素化和降解54。
凋亡的诱导可能不是TNFα的主要生理作用。 TNFα通过激活转录因子,NF-κB和c-Jun介导炎症反应并调节免疫功能。 TNFα的不适当产生或TNFR信号的持续激活与多种人类疾病的发病机制有关,包括自身免疫性疾病,如多发性硬化症55、56和类风湿性关节炎57。事实上,抗TNFα治疗已成为一种有效的方法。这些自身免疫性疾病的治疗。
在信号通路 哺乳动物细胞凋亡的线粒体途径集中于一个关键事件:线粒体外膜通透性(MOMP),被认为是细胞凋亡诱导中的不归路。由于MOMP,某些蛋白质从线粒体膜间空间释放会触发caspase激活级联反应,导致不可逆转的事件最终导致细胞凋亡。 通常由Bcl-2家族的抗凋亡成员阻止MOMP,该成员由抗凋亡蛋白和促凋亡蛋白组成。 Bcl-2首先被描述为B细胞淋巴瘤中独特的致癌基因,可抑制细胞死亡58、59、60。自从鉴定Bcl2以来,已经发现了许多家族成员。这些蛋白质最多共享四个Bcl-2同源域(BH1至BH4)。根据Bcl-2家族在细胞凋亡中的作用及其共享的BH区,通常将其分为三个亚组:一个抗凋亡组和两个促凋亡组。抗凋亡基团包含共享三个或四个BH区的Bcl-2,Bcl-xL61,Bcl-w 62,Bcl-B 63,A1 64和Mcl-1 65。一组促凋亡的Bcl-2家族成员,包括Bax 66,Bak 67、68,Bcl-xs61 Bok 69和Bcl-GL 70,具有两个或三个BH域。另一组包含仅BH3结构域的仅BH3蛋白,包括Bad 71,Bid 72,Bim 73,Bik 74,Noxa 75,Puma 76,Bcl-Gs 70,Blk 77,Bmf 78和Hrk 79。 在促凋亡蛋白中,Bax和Bak似乎是MOMP所必需的,并且很可能通过在线粒体外膜80上形成大小不确定的开口来直接介导MOMP。缺乏Bax和Bak的细胞对许多凋亡刺激具有抵抗力,通过线粒体破坏诱导细胞死亡81、82。Bax和Bak以非活性形式存在于大多数细胞中,它们的活化直接或间接地被其他蛋白质(例如仅BH3的蛋白质)触发。一旦激活,Bax和Bak就会渗透线粒体的外膜,从而导致促凋亡因子(例如细胞色素c)的释放。
Bax和Bak的这种激活被抗凋亡的Bcl-2家族蛋白抑制,后者抑制凋亡激活的仅BH3的蛋白,或者隔离Bax和Bak以阻止其激活。抗凋亡的Bcl-2蛋白对Bax,Bak的这种抑制作用可以通过仅BH3的蛋白以及抗凋亡蛋白的蛋白修饰(例如磷酸化或脱酰胺化)来逆转。 仅BH3蛋白可作为凋亡刺激物的传感器83。在刺激(如细胞因子被剥夺)后,仅BH3蛋白被激活。仍然只有BH3的蛋白直接激活Bax和Bak还是84、85激活它们,还是通过与通常隔离Bax和Bak 86、87的抗凋亡Bcl-2蛋白结合而间接激活Bx和Bak,仍然是一个备受争议的问题。 一旦发生MOMP,膜间空间中的蛋白质就会释放到细胞质中。一种这样的蛋白质,细胞色素c 88,在其WD40结构域结合于胞质单体凋亡蛋白酶活化因子1(APAF-1)。与细胞色素c的这种相互作用会诱导APAF-1发生构象变化,从而促进APAF-1寡聚化,从而启动凋亡小体形成过程26、89、90。然后,凋亡小体通过caspase 9的形式与内在途径的启动子caspase结合。 caspase募集结构域同时存在于APAF-1和caspase 9中。凋亡前体上caspase 9的寡聚化触发了自动裂解,可能是通过使caspase 9分子简单接近而产生了活性caspase 9。最近,发现乙酰胆碱酯酶在细胞凋亡91中很重要,因为它在凋亡小体92、93的形成中起关键作用。然后,活性胱天蛋白酶9裂解效应胱天蛋白酶,例如胱天蛋白酶3和胱天蛋白酶7,从而激活它们。最近,发现细胞内核苷酸可以结合细胞色素c并防止凋亡小体的形成。为了进行细胞凋亡,细胞可能需要打破这种核苷酸屏蔽。 94.尚不知道如何实现。但是,细胞也有可能在不破坏核苷酸屏蔽的情况下发生凋亡。已发现尽管线粒体凋亡95需要APAF-1,但caspase 9激活并不总是需要细胞色素c和凋亡小体,因为caspase 9也可以在带有细胞色素c的小鼠中被激活,而细胞色素c缺乏凋亡诱导功能。 96。MOMP下游的凋亡过程在凋亡小体的水平以及随后激活的每个胱天蛋白酶的水平受到调节。胱天蛋白酶活性可以通过凋亡蛋白抑制剂(IAPs)97、98家族的胱天蛋白酶结合蛋白来调节。例如,人X连锁IAP(XIAP)至少直接抑制胱天蛋白酶3和7 99。在许多人类肿瘤中观察到了存活分子,包括XIAP 98和幸存分子100。此外,这些半胱天冬酶抑制剂可被促凋亡蛋白,具有低pI的半胱天冬酶/直接IAP结合蛋白的第二线粒体衍生激活剂(SMAC / DIABLO)101、102拮抗。SMAC/ DIABLO可取代XIAP与其相互作用激活caspase 3,使细胞死亡继续进行。因此,可以通过多种结合蛋白调节胱天蛋白酶的激活和功能。
在凋亡中起调节作用的另一线粒体成分是凋亡诱导因子(AIF)103。AIF与细菌氧化还原酶具有同源性,通常位于线粒体中,但在凋亡过程中易位至细胞质。 caspase抑制剂不能抑制AIF诱导的细胞凋亡,并且可以导致核浓缩和分裂,而与其他促凋亡因子无关。有趣的是,AIF的氧化还原活性酶区具有抗凋亡作用,使该分子在细胞存活和细胞死亡中具有双重作用。对半胱天冬酶非依赖性凋亡的详细途径了解甚少。
为了描述起见,使用内在和外在途径。实际上,这些途径之间存在串扰。根据细胞类型和刺激以及其他环境因素,不同的凋亡途径发挥不同的作用。例如,在没有足够量的活化半胱天冬酶8允许直接裂解和激活效应半胱天冬酶的细胞中,半胱天冬酶8裂解仅BH3蛋白的Bid可能成为主要途径(图1)。 Bid的这种切割产生了促凋亡的tBID片段,该片段诱导了细胞色素c从线粒体的释放和caspase 9的激活104、105。caspase 9随后可以切割caspase 8,从而形成一个正反馈回路,放大了来自caspase 8的原始信号。
其他凋亡途径
粒酶形成不同于胱天蛋白酶的丝氨酸蛋白酶家族,其也诱导细胞死亡。 T细胞和自然杀伤细胞利用颗粒胞吐途径消除病毒感染的细胞。细胞毒性颗粒通过尚未确定的途径将穿孔素和颗粒酶传递至靶细胞。每个颗粒酶物种触发的凋亡信号传导过程相对不同。颗粒酶B通过胱天蛋白酶依赖性和非依赖机制106触发凋亡,所述机制涉及下游胱天蛋白酶底物的直接切割。颗粒酶B已显示可裂解caspase 3,caspase 8 18和其他底物,包括Bid和ICAD(caspase激活的脱氧核糖核酸酶的抑制剂),可导致CAD内切核酸酶的激活。粒酶A通过诱导单链DNA缺口,通过不依赖caspase的途径诱导细胞死亡。它靶向SET复合物(270至420kDa内质网相关复合物),导致所选成分降解,释放NM23-H1 DNase,并产生单链DNA缺口107。最近显示出颗粒酶可诱导细胞凋亡。在容纳它们的牢房中108。
另一个介导凋亡的半胱氨酸蛋白酶家族-钙蛋白酶(钙激活的中性蛋白酶)是非溶酶体细胞内半胱氨酸蛋白酶。哺乳动物的钙蛋白酶包括两种普遍存在的蛋白,CAPN1和CAPN2,两种胃特异性蛋白,以及具有肌肉特异性的CAPN3。钙蛋白酶还与胱氨酸蛋白酶109共享一些共同的底物。此外,纯化的钙蛋白酶以钙依赖的方式裂解Bax110。已经表明中性粒细胞需要钙蛋白酶来激活BAX和凋亡111、112。最近,已经表明胱天蛋白酶与钙蛋白酶113、114之间的相互作用。进一步阐明蛋白酶诱导的细胞死亡应该揭示不依赖胱天蛋白酶的程序。
淋巴细胞凋亡
T细胞的凋亡在其发育,分化和体内平衡中起着关键作用。几种机制参与控制T细胞不同亚群的凋亡。即使是相同的子群体,在不同情况下也可以使用不同的机制。
胸腺内选择
长期以来,已经表明凋亡在T细胞的胸腺内选择过程中起着至关重要的作用。一般认为,未成熟的T细胞一旦识别出自身MHC分子呈递的自身抗原,就会发生凋亡,这一过程称为负选择115。尽管负选择一直是许多重要研究的重点,但控制负选择的分子机制仍然很大程度上未知。
死亡受体的作用已在各种细胞系统中确立,但是死亡受体是否在胸腺中T细胞的负选择中起作用仍存在争议。 TRAIL-/-小鼠表现出未成熟胸腺细胞数量增加,再刺激116诱导的活化T细胞凋亡减少,同时也有证据表明TRAIL-/-小鼠淋巴或骨髓细胞稳态或T细胞功能没有缺陷117、118此外,由于抗CD3刺激不能被可溶性DR5-Fc 119阻断,因此体外抗CD3刺激诱导的胸腺细胞死亡是不依赖TRAIL的,尽管已证明TNFR1 120、121,TNFR2 122或TNFR1和TNFR2双123缺陷小鼠和Fas / FasL缺陷小鼠124、125、126、127、128表现出正常的阴性选择,在某些模型129、130中也有证据支持TNF和Fas / FasL在阴性选择中的作用例如,小鼠中的TNF过表达导致小胸腺,双阳性胸腺细胞131数量减少。然而,在缺乏FADD或半胱天冬酶8 132、133的小鼠中,负选择是完整的。 ath受体信号转导,提示死亡受体在阴性选择中不重要。但是,仍需要进一步的系统研究以阐明死亡受体在阴性选择中的作用,并且由于使用不同的阴性选择模型有时会导致得出不同的结论123,因此应该对模型进行标准化。
激活诱导的细胞死亡
AICD 134、135、136维持外周T细胞的稳态,尤其是在被特定抗原激活后。稳态的CDD在控制免疫反应的活力和持久性方面起着关键作用。已显示AICD主要由外部Fas / FasL途径137、138、139、140介导,因为可溶性Fas-Fc融合蛋白或中和抗FasL抑制AICD以及Fas和FasL干扰Fas / FasL相互作用TCR结扎后不久被上调。即使阻断了Fas / FasL信号传导,一些T细胞也确实会发生凋亡,但这表明,Tas 138的AICD也参与了Fas非依赖性途径。来自艾滋病患者的T细胞中的AICD依赖于TRAIL。并不是被Fas或FasL 141的抗体所阻断,而是被TRAIL 142的中和抗体所阻断。但是,也有相互矛盾的数据显示,活化后外周T细胞中TRAIL受体的表达上调,但是这些细胞仍然对TRAIL 119具有抗性。一些研究也已经暗示在AICD 126、143、144中起作用。发现TCR刺激可以诱导TNF 145的表达,并且针对TNF的抗体可以部分抑制抗原诱导的体内T细胞的细胞死亡126。另外,针对TNF的抗体的微注射抑制了CD8细胞143中由抗CD3诱导的AICD。然而,也已经报道了TNFR:Fc融合蛋白不抑制效应T细胞146的AICD。
我们上面提到的AICD主要是在来自病原体或抗原引发动物的大量淋巴细胞(未纯化的淋巴细胞,而不是纯化的淋巴细胞亚群)或纯淋巴细胞亚群中进行研究的。可以想象,在不同类型的淋巴细胞中和在不同情况下诱导的AICD通过不同的途径介导。在过去的二十年中,对淋巴细胞的AICD进行了大量的研究。有关淋巴细胞AICD的途径存在一些差异,这主要是由于所用AICD的模型不同所致。例如,抗原或抗体,刺激和再刺激的持续时间和剂量因实验而异。淋巴细胞AICD涉及的确切机制有待进一步研究。将来,在正常或疾病情况下,可能会使用一些标准化模型来剖析参与淋巴细胞AICD的凋亡途径。在以下段落中,我们将讨论一些分化的效应T细胞亚组中的AICD。这些细胞在相对标准化的特异性细胞因子环境下分化。这些效应细胞在AICD中采用的途径有时不同于大细胞AICD中所见的途径。另外,不同效应细胞中的AICD通过不同机制执行(表1)。
CD4 + T细胞根据活化过程中的细胞因子环境,分为I型(Th1)和II型(Th2)辅助T细胞。Th1细胞进化为清除细胞内病原体,并根据其产生的干扰素进行定义-γ(IFN-γ)。另一方面,Th2细胞对于控制某些寄生虫感染至关重要,并由其细胞因子IL-4,IL-5和IL-13的产生来定义。虽然尚不清楚Fas / FasL相互作用是否参与负选择,但可以肯定的是Fas / FasL有助于Th1 149、150的极化。Th1细胞易受AICD感染。它们在抗原刺激后经历快速凋亡。 Th1细胞中的AICD主要由Fas / FasL介导。与来自正常小鼠的Th1细胞相比,从gld或lpr小鼠产生的Th1细胞没有显示出快速的AICD。可溶性Fas可能会阻断Th1细胞中的AICD。当用抗原刺激时,Th2细胞也经历AICD。但是,它们死亡的速度要慢得多,可溶性Fas对Th2细胞中AICD的作用有限。与Th1细胞不同,Th2细胞对Fas介导的细胞凋亡具有抗性151,152;但是,其潜在机制存在争议。一些研究表明,Th2细胞表达的Fas / FasL较少150、153。另一方面,也有证据表明Th1和Th2细胞之间Fas / FasL的表达没有差异154。此外,还发现Th2细胞表达的Fas / FasL表达较高。 FAP-1(一种与Fas相关的磷酸酶可能起抑制Fas信号转导的作用)水平154。但是,还有其他数据显示,TCR激活诱导的Th2细胞中磷脂酰肌醇3激酶活性的选择性上调抑制了caspase 8的裂解。认为是Th2抵抗Fas介导的细胞凋亡的机制155,156。 与Th1细胞相比,对Th2细胞中的AICD的研究较少。长期以来,尚不清楚什么分子负责Th2细胞中的AICD。最近,人们发现粒酶B发挥着关键作用157。死亡受体不参与Th2细胞的AICD,因为阻断它们的同源配体对活化的Th2细胞的凋亡没有影响。但是,抑制粒酶B活性消除了Th2细胞中的AICD。此外,源自缺乏粒酶B的小鼠的Th2细胞对AICD具有抗性。结果,粒酶B缺乏或粒酶B活性的抑制增加了Th2细胞因子的产生并增加了对变应原诱导的哮喘的易感性。
Th17(也称为Thi)是分泌IL-17的效应子T细胞的新亚组,最近已被描述为158、159,并引起了人们的极大兴趣。 Th17细胞已显示在某些免疫相关疾病的发病机理中起重要作用。因此,研究这种关键的T细胞群体如何经历AICD将会很有趣。最近,我们发现抗Fas的中和抗体在Th17细胞中很大程度上阻断了抗CD3诱导的AICD,而阻断TRAIL和TNF则未显示任何作用(张英宇和徐光武等,正在准备论文)。这些结果表明,Fas / FasL可能是Th17细胞AICD参与的主要途径。 Fas / Fas配体系统参与了CD4 + T细胞的AICD,但对于CD8 + T细胞死亡160似乎不是必需的。相反,通过TNFR的凋亡信号可能参与了CD8 + T细胞143、161的AICD。在缺乏TNFR p55 161的小鼠中观察到功能激活的细胞毒性T细胞的持久性。在这项旨在了解CD4 + T细胞帮助在CD8 + T细胞记忆中的作用的研究中,记忆CD8 + T细胞没有适当的CD4 + T细胞帮助,二次刺激后接受AICD。这些无助的CD8 + T细胞通过介导AICD 162的抗原再刺激合成TRAIL。但是,应该指出,记忆CD8 + T细胞是分化的CD8 + T细胞。这些细胞中的AICD可能与原始CD8 + T细胞或体外分化的Tc细胞中的AICD不同。最近,有报道说,RNA干扰消除了粒酶B,从而抑制了蛋白酶3的加工和tBid的产生,并增强了CD3刺激的CD8 + T细胞的存活。此外,这种作用与穿孔素状态无关。163.综合考虑,构成CD8 + T细胞AICD的主要途径的构成问题尚待进一步研究。
与它们的CD4 +对应物相似,已经在小鼠164、165、166,大鼠167、168和人169、170、171中建立了CD8 + T细胞不同亚群的存在。它们被称为Tc1和Tc2。 Tc1细胞分泌IFNγ,而Tc2细胞分泌IL-4,IL-5,IL-6和IL-10164。关于AICD在Tc1和Tc2中的数据有限。像Th1细胞一样,Tc1细胞也比2型对应物更易受AICD的侵扰,而Fas / FasL在Tc1细胞的AICD中起着重要作用,因为针对FasL的中和抗体部分废除了AICD。但是,已发现Granzyme B抑制剂(z-AAD-CMK)也抑制Tc1细胞中的AICD,表明Tc1 AICD 146参与了颗粒胞吐机制。在缺乏Granzyme B的小鼠中进行进一步的实验将有助于理解其作用。 Tc1 AICD中的粒酶B序列。尽管Tc2对Fas / FasL诱导的细胞凋亡具有相对抗性,但在两个子集146上均发现Fas和FasL的细胞表面表达水平相似。尚不清楚为什么Tc2对Fas / FasL诱导的细胞凋亡具有相对抗性,以及AICD如何在Tc2细胞中受到调控。 T细胞其他亚群(例如NKT细胞和CD4 + CD25 +调节性T细胞)采用的凋亡机制尚不清楚。然而,这些细胞似乎对凋亡172、173具有相对抗性。新鲜分离的调节性T细胞对CD95介导的凋亡高度敏感,但在TCR重新刺激后,与CD4 + CD25−相比,调节性T细胞对AICD的敏感性降低。 T细胞174.由于这些细胞在先天免疫和免疫调节中很重要,因此研究它们的AICD将为理解免疫系统提供新的信息。 与我们对T细胞中AICD的了解相比,对B细胞凋亡的了解要少得多。像在T细胞中一样,通过B细胞受体激活而诱导的细胞死亡也被称为AICD 175、176。然而,与T细胞不同,B细胞中的AICD几乎与死亡受体无关,而是取决于死亡受体。内在途径177.通过诸如FADD / MORT1或CrmA之类的抑制剂阻断Fas / FasL系统或阻止FADD信号传导,并未阻断B细胞中的AICD。此外,Fas缺陷型lpr小鼠表现出BCR结扎诱导的B细胞死亡,与野生型178、179、180相似。BCR交联导致线粒体形态发生变化,包括线粒体膜的广泛破坏和在此期间线粒体的肿胀BCR诱导的细胞凋亡181;它也导致线粒体膜电位182、183、184的去极化。此外,可以稳定线粒体膜电位的线粒体抑制剂能够保护B细胞免受AICD 181、185的侵害。仍然不清楚B细胞AICD中线粒体信号传导调控的许多细节。
控制各种淋巴细胞群体凋亡的机制明显不同。这些差异已开始被人们所认识,并引起了免疫学家的关注。毫无疑问,在这一激动人心的领域的进一步研究将揭示出新颖的信息,这些信息将增进我们对免疫系统及其调节机制的理解。
Apoptosis signaling pathways and lymphocyte homeostasis | Cell Research
https://www.nature.com/articles/cr200752
e selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells Original Paper Published: 17 July 2003 The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells A Troyano, P Sancho, C Fernández, E de Blas, P Bernardi & P Aller Cell Death & Differentiation volume 10, pages889–898(2003)Cite this article 454 Accesses 79 Citations 1 Altmetric Metrics details Abstract Treatment with 0.2 mM hydrogen peroxide (H2O2) or with 0.5 mM cisplatin caused caspase-9 and caspase-3 activation and death by apoptosis in U-937 human promonocytic cells. However, treatment with 2 mM H2O2, or incubation with the glutathione suppressor DL-buthionine-(S,R)-sulfoximine (BSO) prior to treatment with cisplatin, suppressed caspase activation and changed the mode of death to necrosis. Treatment with 2 mM H2O2 caused a great decrease in the intracellular ATP level, which was partially prevented by 3-aminobenzamide (3-ABA). Correspondingly, 3-ABA restored the activation of caspases and the execution of apoptosis. By contrast, BSO plus cisplatin did not decrease the ATP levels, and the generation of necrosis by this treatment was not affected by 3-ABA. On the other hand, while all apoptosis-inducing treatments and treatment with 2 mM H2O2 caused Bax translocation from the cytosol to mitochondria as well as cytochrome c release from mitochondria to the cytosol, treatment with BSO plus cisplatin did not. Treatment with cisplatin alone caused Bid cleavage, while BSO plus cisplatin as well as 0.2 and 2 mM H2O2 did not. Bcl-2 overexpression reduced the generation of necrosis by H2O2, but not by BSO plus cisplatin. These results indicate the existence of different apoptosis/necrosis regulatory mechanisms in promonocytic cells subjected to different forms of oxidative stress.
The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells
Original Paper
Published: 17 July 2003
The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells
A Troyano, P Sancho, C Fernández, E de Blas, P Bernardi & P Aller
Cell Death & Differentiation volume 10, pages889–898(2003)Cite this article
Abstract
Treatment with 0.2 mM hydrogen peroxide (H2O2) or with 0.5 mM cisplatin caused caspase-9 and caspase-3 activation and death by apoptosis in U-937 human promonocytic cells. However, treatment with 2 mM H2O2, or incubation with the glutathione suppressor DL-buthionine-(S,R)-sulfoximine (BSO) prior to treatment with cisplatin, suppressed caspase activation and changed the mode of death to necrosis. Treatment with 2 mM H2O2 caused a great decrease in the intracellular ATP level, which was partially prevented by 3-aminobenzamide (3-ABA). Correspondingly, 3-ABA restored the activation of caspases and the execution of apoptosis. By contrast, BSO plus cisplatin did not decrease the ATP levels, and the generation of necrosis by this treatment was not affected by 3-ABA. On the other hand, while all apoptosis-inducing treatments and treatment with 2 mM H2O2 caused Bax translocation from the cytosol to mitochondria as well as cytochrome c release from mitochondria to the cytosol, treatment with BSO plus cisplatin did not. Treatment with cisplatin alone caused Bid cleavage, while BSO plus cisplatin as well as 0.2 and 2 mM H2O2 did not. Bcl-2 overexpression reduced the generation of necrosis by H2O2, but not by BSO plus cisplatin. These results indicate the existence of different apoptosis/necrosis regulatory mechanisms in promonocytic cells subjected to different forms of oxidative stress.A factor that may trigger cell death, and that may decide the selection between apoptosis and necrosis, is the increase in intracellular oxidation. Using treatment with exogenous hydrogen peroxide (H2O2) as an experimental paradigm of oxidation, it was often observed that moderate H2O2 concentrations triggered apoptosis, while elevated concentrations caused necrosis (the exact concentrations depending very much on the used cell model).5,9,10,11 Moreover, the application of a low concentration of exogenous H2O2 sufficed to suppress apoptosis and cause necrosis-like death in Burkitt's lymphoma cells treated with antitumour drugs.11 In this model, the change in the mode of death was attributed to the oxidant-mediated depletion of intracellular ATP, which is a strict requirement for the execution of apoptosis. However, excessive oxidation might also suppress apoptosis by other mechanisms, such as direct inactivation of caspases.10,12 An alternative method to cause oxidation is by decreasing the intracellular concentration of antioxidant molecules, making the cell unable to cope with the normal production of reactive oxygen species (ROS). In this manner, the depletion of intracellular reduced glutathione (GSH) by prolonged incubation with DL-buthionine-(S,R)-sulfoximine (BSO), a specific inhibitor of γ-glutamylcysteine synthetase, caused an increase in the intracellular content of H2O2, and potentiated the increase provoked by other treatments.13,14 Correspondingly, the incubation with BSO potentiated the lethality of cytotoxic treatments,13,14,15 leading in some cases, to a change of the mode of death from apoptosis to necrosis.7,16
We have recently reported that the alkylating drug cis-platinum(II)-diammine dichloride (cisplatin, CDDP), which normally caused death by apoptosis, provoked necrosis when applied to U-937 promonocytic cells with a reduced GSH content.8 Although necrosis induction was accompanied by an increased intracellular concentration of peroxides, and could be prevented by antioxidants, it remained unclear whether the change in the mode of death could be adequately explained as the simple consequence of increased ROS accumulation. As a further investigation, in the present work we comparatively analyse some mechanisms critical for the regulation of cell death in U-937 cells subjected to the above-described models of oxidation, namely treatment with exogenous H2O2, and treatment with cisplatin with and without GSH suppression. The results demonstrate that, among other differences, the switch from apoptosis to necrosis in H2O2-treated cells is the consequence of ATP depletion, while in BSO plus cisplatin-treated cells it is independent of ATP, and apparently associated with the inhibition of mitochondrial cytochrome c release.
ATP levels
Apoptosis is an energy-dependent process, in such a manner that the decrease of ATP below critical levels may impede the execution of apoptosis and promote necrosis.19,20 For this reason, experiments were carried out to compare the fluctuations of ATP levels upon treatment with H2O2 and cisplatin, both under apoptosis and necrosis-inducing conditions. The treatments were carried out for a maximum of 3 h, to prevent possible ATP leakage through the damaged plasma membranes in necrotic cells. The results are indicated in Figure 3. Treatments with 0.2 mM H2O2 and with cisplatin alone caused a slight decrease in the ATP level, which at 3 h reached levels of 60–75% of the control value. Treatment with 2 mM H2O2 caused a rapid and profound depletion of ATP, which at 1 h reached levels of around 10% in relation to control cells. By contrast, BSO plus cisplatin did not decrease, and even augmented the ATP content.Cytochrome c release
The release of the cytochrome c from the mitochondria to the cytosol is required for the assembly of the apoptosome and hence for the activation of the caspase cascade in the intrinsic pathway of apoptosis.17
Expression of Bcl-2 protein family members
The release of cytochrome c is regulated by proteins of the Bcl-2 family, which may either inhibit (e.g. the antiapoptotic proteins Bcl-2 and Bcl-XL) or promote (e.g. the proapoptotic protein Bax) the process.27 Bax is normally present as an inactive monomer in the cytosol, and to be functional requires oligomerisation and translocation to the mitochondrial membrane.27 In addition, cisplatin and other cytotoxic drugs may stimulate Bid cleavage and insertion into the mitochondrial membrane, where it also promotes cytochrome c release.28,29 For these reasons, immunoblot assays were carried out to measure the level, integrity and subcellular distribution of Bcl-2, Bcl-XL, Bax and Bid. The results, indicated in Figure 6, were as follows: (i) Bcl-2, Bcl-XL and Bax were constitutively expressed in untreated cells, and their total levels were little affected by treatment with H2O2 and cisplatin, both under apoptosis- and necrosis-inducing conditions (Figure 6a). (ii) In untreated cells, Bax was mostly detected in the cytosolic fraction and only a minimal part in the mitochondrial fraction. Treatment with 0.2 and 2 mM H2O2 and with cisplatin alone, which as indicated above provoked cytochrome c release, caused a progressive increase (from 2 to 3 h) in Bax levels in mitochondrial extracts. By contrast, there was no increase in mitochondrial extracts from cells treated with BSO plus cisplatin, which failed to cause cytochrome c release (Figure 6b). Bcl-2 was always undetectable in cytosolic fractions (result not shown), and its level in the mitochondrial fractions was not significantly modified by the treatments, being used as an internal control (Figure 6b). (iii) Treatment with cisplatin alone caused a progressive loss (from 2 to 3 h) of Bid proform (21 kDa), while treatments with BSO plus cisplatin and with 0.2 and 2 mM H2O2 did not (Figure 6c). In spite of the commercial specifications of the used antibodies, it was almost impossible to detect Bid fragments by Western blot. Hence, we consider the loss of Bid proform as an indication of cleavage, a criterion also used by other authors.30,31
The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells | Cell Death & Differentiation
https://www.nature.com/articles/4401249
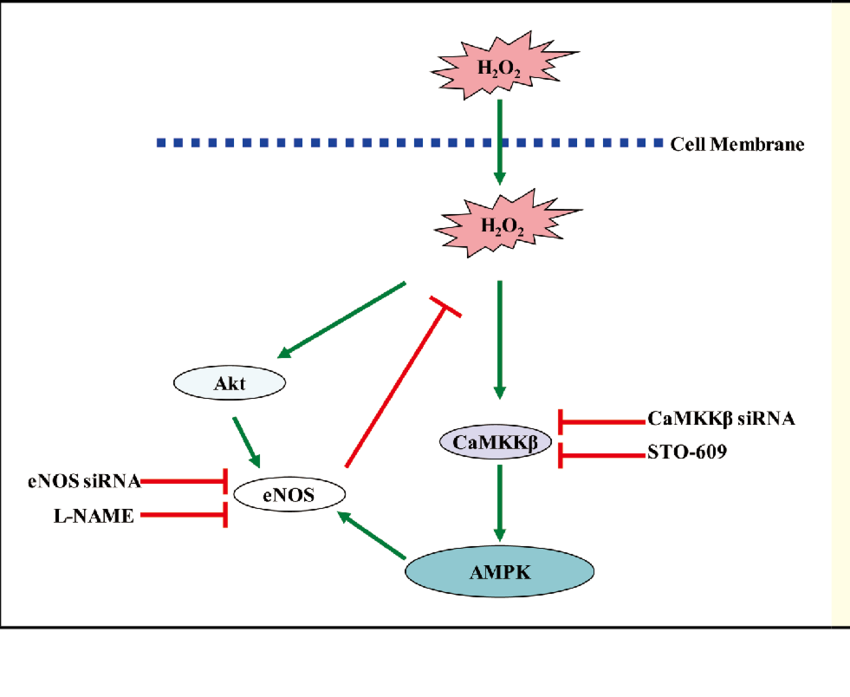
Model for eNOS regulation of CaMKKβ/AMPK pathways via H2O2. Genetic or pharmacological suppression of the eNOS-NO pathway increases intracellular H2O2 levels and thereby leads to CaMKKβ/AMPK-dependent phosphorylation of AMPK. In this model, H2O2 directly or indirectly activates CaMKKβ. The phosphorylated AMPK, in turn, phosphorylates and activates eNOS, representing a feedback mechanism controlling this pathway. AMPK, AMP-activated protein kinase; eNOS, endothelial nitric oxide synthase. (Adapted with modifications from Jin et al. 101 )
Model for eNOS regulation of CaMKKβ/AMPK pathways via H2O2. Genetic or... | Download Scientific Diagram
https://www.researchgate.net/figure/Model-for-eNOS-regulation-of-CaMKKb-AMPK-pathways-via-H2O2-Genetic-or-pharmacological_fig6_232280199
.png)
.jpg)